Article Text

Statistics from Altmetric.com
The interrelation of neurology and the gastrointestinal system includes defects of gut innervation, primary disorders of the nervous system (or muscle) which lead to gastrointestinal symptoms—for example, dysphagia—and, finally, certain gut disorders which include neurological features in their clinical range. The first of this trio will be discussed only briefly in this review, the second and third in more detail.
Defects of innervation
ACHALASIA
Achalasia is characterised by an absence of peristalsis in the oesophageal body accompanied by a failure of relaxation of the lower oesophageal sphincter.1 Although the condition can be secondary to other disease processes—for example, Chagas’ disease—in Europeans it is usually a primary disorder. Differing opinions have been expressed as to whether the problem of innervation rests in the dorsal motor vagal nucleus, the vagus itself, or in the intrinsic innervation of the oesophagus, with most evidence favouring the last explanation. By the time of oesophageal biopsy or resection, there is almost total loss of ganglion cells with substantial destruction of myenteric nerves. The changes are accompanied by an inflammatory reaction both within and around the nerves. Neurochemical analysis has shown a reduction in the number of neurons in the myenteric plexus containing immunoreactive vasoactive intestinal polypeptide.2 The way in which the disease evolves remains unclear.
HIRSCHSPRUNG’S DISEASE
Hirschprung’s disease presents at, or soon after, birth. Constipation is accompanied by gaseous abdominal distension. Typically a narrowed distal segment of bowel is demonstrable in which there is loss of parasympathetic ganglion cells from the intramural plexus.3 The aganglionosis is the result of incomplete migration of neurenteric ganglion cells from the neural crest to the most distal part of the gut. Increased acetylcholinesterase activity has been detected in the submucosal and myenteric plexus of the affected bowel segment. Besides using histological criteria for diagnosis—namely, the presence or absence of ganglion cells in rectal biopsy4—acetylcholinesterase activity can be measured in the same specimen.5 Further experience has established that the two techniques are complementary, acetylcholinesterase staining being particularly helpful when the biopsy material does not include submucosa, or in older infants or children in whom the population of distal submucosal ganglion cells may be less dense.6
Gastrointestinal disorders due to neurological disease
DYSPHAGIA
A neurogenic mechanism for dysphagia, which may have either sensory or motor components, or both, can result from a disorder at the oral, pharyngeal, or oesophageal phase of swallowing. In most patients, the neurological disorder is evident, but in others, dysphagia is the presenting feature. Besides the dysphagia, other symptoms suggesting a neurogenic mechanism include drooling of saliva, nasal regurgitation, and episodes of coughing or choking during swallowing.7Videofluoroscopy has proved of particular value in the assessment of neurogenic dysphagia. The procedure allows identification of the site of maximal dysfunction, pinpoints areas of barium collection, and indicates whether laryngeal penetration is occurring.8Neurogenic dysphagia may arise from involvement of the cortical areas concerned with swallowing, their efferent pathway, the brain stem motor or sensory nuclei, the lower cranial nerves in their distal course, their neuromuscular junctions, or the striated muscle components of the swallow pathway.
STROKE
Stroke is the commonest cause of neurogenic dysphagia. Up to 50% of patients with stroke have been estimated to have dysphagia, albeit temporary in many. Dysphagia is a recognised feature of unilateral as well as bilateral hemispheric stroke and is commonplace in brain stem stroke. Most studies of dysphagia in cases of unilateral hemispheric stroke have been retrospective, but in one prospective study, swallow function was analysed with respect to the size and distribution of ischaemic stroke in middle cerebral artery territory.9Attempts to correlate swallow patterns with stroke site were hampered by the fact that stroke volumes for lesions in the anterior territory of the middle cerebral artery were substantially larger than those in the posterior territory of the artery. Pharyngeal transit time was prolonged, compared with controls, with lesions in either hemisphere. Laryngeal penetration and aspiration were much more common in the right hemisphere group. In general, however, attempts to correlate characteristics of unilateral lesions with impairment of swallowing have not produced consistent findings. Data from experimental animals has suggested that stimulation of either cortex can initiate swallowing.10 Transcranial magnetoelectric stimulation has been used to study the projections of the corticofugal fibres involved. The oral muscles are represented symmetrically between the two hemispheres, whereas muscles of the pharynx and oesophagus tend to be represented asymmetrically, but without regard to speech dominance.11 The technique has been applied to the analysis of patients with unilateral hemispheric stroke, with or without dysphagia.12 In patients with dysphagia, pharyngeal responses from the unaffected hemisphere are smaller than those in non-dysphagic patients, irrespective of the side of the lesion, or whether it is cortical or subcortical. The mylohyoid responses (taken as representative of oral swallowing musculature) do not display such asymmetry. It has been suggested that this implies that pharyngeal function is represented asymmetrically in the cortex, and that with damage to the hemisphere containing the predominant pharyngeal centre, swallowing function cannot be maintained by the “non-dominant” hemisphere. Clearly, if this hypothesis is correct, an alternative mechanism for dysphagia must exist in the small proportion of patients with a predominant oral phase disorder of swallowing after hemispheric stroke.
Some degree of swallow difficulty is remarkably common after unilateral hemispheric stroke. It was reported in nearly 30% of one series, based on the bedside assessment of swallowing liquid.13 Some evidence was found for an adverse effect on functional outcome if dysphagia was present. By one month after onset of stroke, only 2% of patients with unilateral stroke are still dysphagic. Clearly any hypothesis regarding the pathogenesis of dysphagia after unilateral stroke needs to explain the transient nature of the process in many patients.
Bilateral hemispheric strokes are associated with a higher incidence and greater severity of dysphagia than unilateral strokes.7 Generally, the neurogenic basis of the problem is evident from the patient’s examination although case reports exist describing dysphagia in patients with occult bilateral hemispheric infarction.14 Swallow problems are particularly common in brain stem stroke, and are likely to include aspiration. Although aspiration is more likely in those with bilateral brain stem lesions, the presence of unilateral or bilateral infarction does not correlate with outcome.15 As with hemispheric stroke resulting in dysphagia, many dysphagic patients with brain stem strokes are able to return to full oral nutrition.
EXTRAPYRAMIDAL DISORDERS
Abnormalities of swallowing described in Parkinson’s disease include defects of tongue movements, a delayed swallowing reflex, aspiration, and reduced pharyngeal peristalsis. Videofluoroscopy has allowed a more detailed analysis, particularly necessary as patients’ symptoms correlate poorly with the type of swallow problem.16 Silent laryngeal aspiration is commonly found. Alhough opinions differ, recent studies indicate that, for some patients, significant improvement in swallow function occurs with medication. Patients with Parkinson’s disease who deny swallowing difficulties have also been studied.17 In such a group of 16 patients, all had some abnormality and three had silent aspiration. Their mean Webster score (used as an assessment of their disability) was 11, indicating relatively mild disease, and assessments were performed at the time of the midday meal.
Dysphagia is a prominent feature of progressive supranuclear palsy18 and is a recognised finding in both Huntington’s and Wilson’s disease. Dysphagia in patients with spasmodic torticollis partly relates to the variable head and neck posture but, in addition, delay in reflex initiation and the finding of pharyngeal residue on videofluoroscopy suggests a neurogenic component.19
OTHER NEUROGENIC DISORDERS
Besides stroke and multiple sclerosis, other brain stem pathologies are associated with dysphagia. In the Chiari type 1 malformation, herniation of the cerebellar tonsils through the foramen magnum results in traction of the lower cranial nerves, secondary compression of the brain stem, and, in some patients, hydrocephalus. Dysphagia is common in such patients and is associated with a global impairment of all phases of swallowing on videofluoroscopy.20 In some patients, dysphagia has been the presenting feature. Palatal hypoaesthesia has usually been the norm, however, when such patients have been carefully assessed.21 22
Disorders of the lower motor neuron or neuromuscular junction that often result in dysphagia include the Guillain-Barré syndrome, amyotrophic lateral sclerosis, and myasthenia gravis. In amyotrophic lateral sclerosis, swallow abnormalities are not infrequent even in those patients presenting with limb problems but are particularly prominent where bulbar involvement is evident on clinical examination. An abnormal oral phase of swallowing encompasses both a prolonged oral transit time and repetitive lingual pumping, probably reflecting reduced lingual force.23 Patients often employ changing head postures to facilitate swallowing. A prolonged delay time (duration from arrival of the bolus at the midpoint of the velum until initiation of maximal hyoid excursion) is strikingly apparent in the bulbar group, whereas the pharyngeal response time (from initiation of maximal hyoid excursion to hyoid return to rest) shows no difference between patients with the bulbar and non-bulbar forms of the disease. Laryngeal aspiration can occur before, during, or after the pharyngeal phase of swallowing.
Dysphagia has been estimated to be a prominent symptom in at least one third of myasthenic patients. Both oral and pharyngeal phases of swallowing may be affected and this despite a lack of subjective complaint of dysphagia.24 In cases with severe bulbar involvement, findings include ballooning of the pharynx during repeated swallowing, residues within the pharynx, nasopharyngeal regurgitation, and poor palatal elevation.
PRIMARY MUSCLE DISEASE
Dysphagia is a recognised feature of inflammatory muscle disease, myotonic dystrophy, and some of the muscular dystrophies. Fluoroscopic features of both polymyositis and dermatomyositis include defective transfer to the oropharynx, retention of material in the valleculae, and aspiration. Peristalsis is defective in the upper oesophagus.25 An additional mechanism for dysphagia in inflammatory muscle disease is failure of relaxation of a dysfunctional cricopharyngeal muscle.26 Rarely, inclusion body myositis presents with dysphagia and, in some of these cases, biopsy of cricopharyngeal muscle has disclosed characteristic features of the disease.27 Cricopharyngeal myotomy can be therapeutically beneficial.
In an early manometric study, evidence was presented of weakness affecting the pharynx, cricopharyngeal sphincter, and oesophagus in myotonic dystrophy, although doubt has been cast on the accuracy of such recording systems.28 In a barium study published in the same year, dilatation of the oesophagus (principally its lower portion) was noted along with diminution and slowing of peristalsis.29 Further video fluoroscopic studies have elaborated on these findings. Abnormalities described include impaired pharyngeal contraction, myotonia of the tongue and pharynx, stasis, and pooling of the contrast in the pyriform sinuses and valleculae along with nasal regurgitation and tracheal aspiration.30 The earlier findings in the oesophagus have been confirmed with, in addition, descriptions of oesophageal spasm, regurgitation, and antiperistaltic contractions. In some patients abnormalities of oesophageal motility are unaccompanied by symptoms.
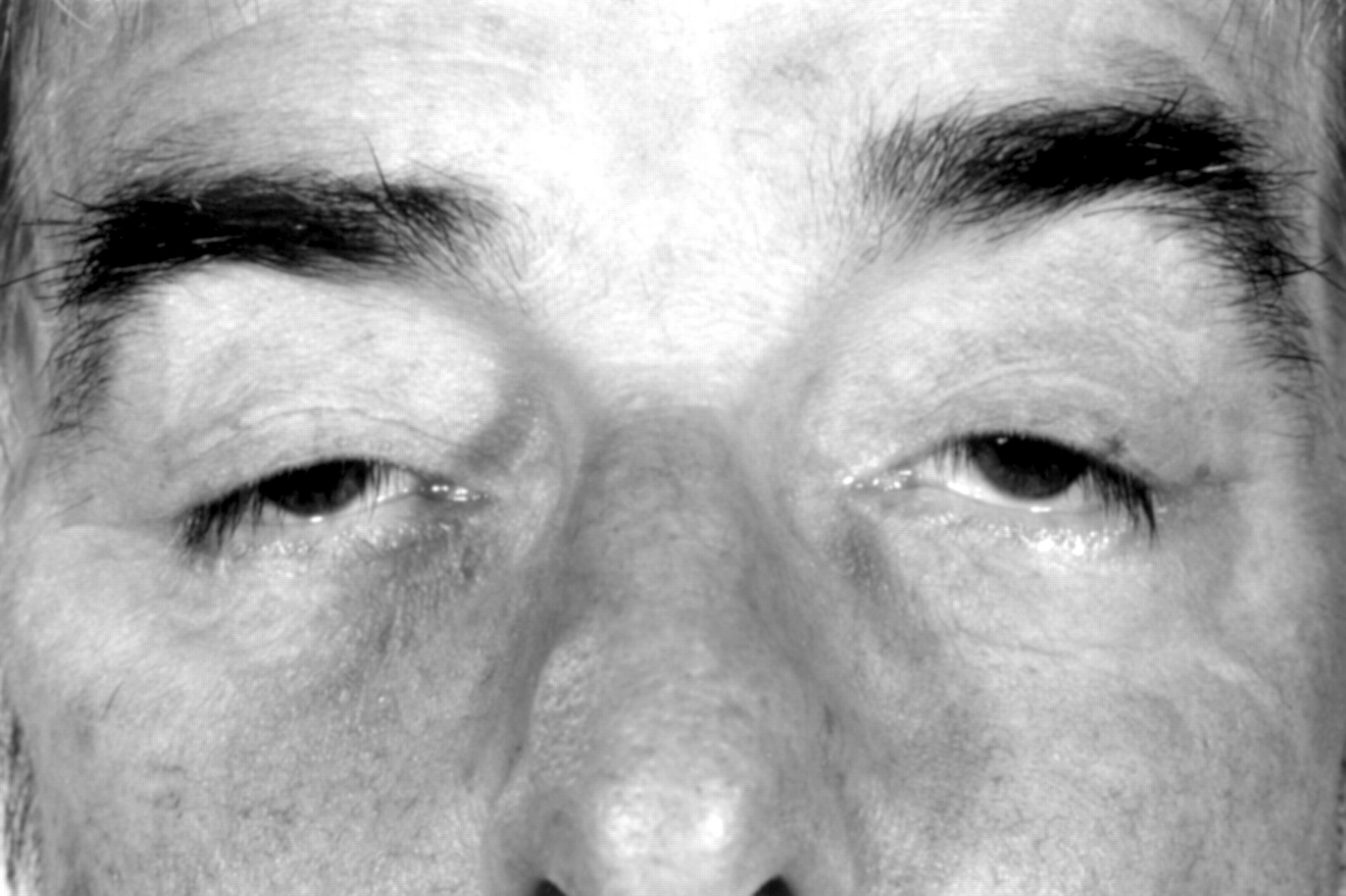
Oculopharyngeal muscular dystrophy is an autosomal dominant disorder which is predominantly seen in French Canadians.31 The condition normally presents with bilateral ptosis, with dysphagia following (fig 1). The ptosis is very variable and sometimes barely discernible. Pharyngeal contraction is typically depressed or absent with pooling in the hypopharynx. Relaxation of the upper oesophageal sphincter is incomplete, late, or absent. Peristaltic activity in the oesophagus is often abnormal. Cricopharyngeal myotomy can result in a significant relief of symptoms. In some families, dysphagia precedes other manifestations of the disease by months or years.32

Bilateral ptosis in a patient with oculopharyngeal muscular dystrophy.
Disorders of gastric and intestinal motility
Both neurogenic and myopathic disorders are associated with abnormalities of gut motility.
NEUROGENIC DISORDERS
With neurogenic disorders involvement of the autonomic innervation of the gut is the relevant mechanism. In this context, the most common underlying disorder is diabetes mellitus. Gastroparesis leads to nausea, vomiting, abdominal pain, and distension. Alhough most patients with a diabetic gastroparesis display antral hypomobility,33 a proportion have periods of continuous, low amplitude, contractions in the antrum, in both the fasting and postprandial phases.34 Abnormalities of the small bowel are common in patients with diabetic gastroparesis. Typically, frequent long and short non-propagated bursts of phasic pressure activity occur in the jejunum with reduced frequency and amplitude of contractions in both the fasting and fed phases.34 The phenomenon of long and short bursts has been attributed to sympathetic denervation and the composite intestinal motility pattern in the fasting phase considered to represent dysfunction of both sympathetic and parasympathetic supply. Both constipation and diarrhoea are common in diabetic patients and indeed may alternate. It has been suggested that the colonic muscle of diabetic patients with constipation is able to respond to exogenous stimulation, in the form of parenteral injection of neostigmine or metoclopramide, but not to the stimulus of an ingested meal.33 Diarrhoea in diabetic patients may be due to gluten induced enteropathy, pancreatic insufficiency, bacterial overgrowth of the small intestine, or so-called idiopathic diabetic diarrhoea. The last has generally been considered a manifestation of autonomic neuropathy. Faecal incontinence is a common problem in patients with diarrhoea.
PRIMARY MUSCLE DISEASE
Abnormal gastric emptying has been identified in patients with polymyositis or dermatomyositis,35 although in most patients it remains asymptomatic. An extensive literature exists on motility disorders associated with myotonic dystrophy (table 1). Delayed gastric emptying is almost inevitable in the condition but is seldom symptomatic.36 Diarrhoea and abdominal cramps are common and steatorrhoea is recorded. Radiological studies of the large bowel have sometimes demonstrated megacolon.37 Intestinal pseudoobstruction has also been reported38 and manometric abnormalities attributable to abnormal and sustained contraction of the internal and external anal sphincters.39 Despite this, constipation is relatively uncommon.
Gastrointestinal manifestations of myotonic dystrophy
Various gastrointestinal symptoms have been recorded in patients with Duchenne’s muscular dystrophy, including diarrhoea and constipation. Cases of intestinal pseudo-obstruction have been encountered with dilated and fluid filled small intestine and colon.40 It has been suggested that in such cases there is significant atrophy and fibrosis of the intestinal smooth muscle.
Primary disorders of gut function or absorption
VITAMIN B12 DEFICIENCY
Accounts of the manifestations of vitamin B12deficiency extend from the last century although, inevitably, early accounts are less likely to have included a homogenous population.
Cobalamin in food is bound to protein. Peptic digestion releases free cobalamin which then binds to R-binder, a cobalophilin found in saliva and gastric juice. Free cobalamin is again formed in the duodenum by the action of pancreatic enzymes but then binds to parietal cell derived intrinsic factor. Absorption takes place via intrinsic factor receptors in the terminal ileum (fig 2).41The primary function of cobalamin is to provide coenzymatic activity for the synthesis of methionine and succinyl-coenzyme A. The mechanism by which reduced methionine causes demyelination in the CNS remains to be elucidated.41 Vitamin B12 deficiency may arise from inadequate oral intake, deficiency of intrinsic factor formation, various malabsorption disorders (for example, in association with jejunal diverticulosis), resection of the stomach or terminal ileum, or from a disorder of the terminal ileum resulting in altered absorption of bound cobalamin. Consequences include peripheral neuropathy, myelopathy, altered mental status, and optic neuropathy.42Patients with pernicious anaemia due to intrinsic factor deficiency are usually in their 60s or 70s at presentation but the condition is recognised in juveniles and young adults. Most patients with pernicious anaemia with neurological dysfunction present with a mixed myelopathic/neuropathic picture. The commonest initial neurological complaint is a mixture of numbness and paraesthesia distributed symmetrically and starting usually either in the feet alone, or in the feet and hands together. In some patients, however, sensory symptoms are confined to the upper limbs. The second commonest presenting complaint, either in isolation or with sensory symptoms, is gait ataxia. Other forms of neurological presentation are unusual. A small proportion of patients have symptoms suggestive of autonomic dysfunction with urinary urgency, frequency, or incontinence, faecal incontinence, or impotence. Typically symptoms progress over weeks or months.

Enteric processing and absorption of cobalamin (Cbl). IF=intrinsic factor; R-binder=A cobalophilin with a rapid (compared with IF) electrophoretic mobility; TCII=transcobalamin II. (This figure is reproduced by kind permission of the author and publisher.41)
The commonest neurological finding is diminished lower limb vibration sense. Sometimes the hands or arms are affected. Proprioception is typically impaired in a similar distribution. Cutaneous sensory change is less frequent. Lhermitte’s sign has been reported in up to 20% of cases.43 Ataxia, commensurate with the impairment of lower limb proprioception, is common. Weakness is sometimes found, but always accompanied by sensory abnormalities, and principally in the lower limbs. There may be lower limb hyperreflexia, or hyporeflexia, or a combination of the two. Hyporeflexia is more common. Visual impairment takes the form of bilateral centrocaecal scotomata. It is uncommon and even more so as an isolated phenomenon.44 Mental impairment usually takes the form of a global dementia or simply a mild impairment of short term memory with a reduced attention span. Neurological investigation provides some assistance in making the diagnosis. Studies with EMG when abnormal have shown evidence of a predominantly axonal neuropathy. Somatosensory evoked potentials may be delayed or absent, with recovery after successful treatment.45 Visual evoked potentials may be abnormal in the absence of visual symptoms or signs.
It is well recognised that a lack of correlation exists between the haematological and neuropsychiatric manifestations of vitamin B12 deficiency. Indeed, severely anaemic patients may display no neurological impairment. Furthermore, some patients may display neuropsychiatric manifestations of B12 deficiency in the absence of anaemia or macrocytosis.46 Most such patients will have evidence of a megaloblastic bone marrow but reports of patients with both normal peripheral blood and bone marrow examinations yet with neuropsychiatric syndromes exist.47
Generally, serum vitamin B12 concentrations are very substantially depressed, although it has been argued that concentrations of 100–200 pg/ml are still consistent with the diagnosis, which is then supported by the finding of raised concentrations of serum methylmalonic acid and total homocysteine and by the response to treatment.46
Other vitamin B group deficiencies
VITAMIN B1 DEFICIENCY
Vitamin B1 deficiency leads to beriberi and Wernicke-Korsakoff syndrome. Although the latter has been intimately linked to chronic alcoholism, it is recognised to occur in other settings. In a postmortem series of 29 cases, in which chronic alcoholism had been excluded, gastrointestinal causes of the syndrome included peptic ulcer, acute pancreatitis, oesophageal metastasis, and carcinoma of the stomach or oesophagus.48 A combination of ophthalmoplegia, nystagmus, and ataxia, considered to reflect a Wernicke-Korsakoff syndrome, has been described in a patient with anorexia nervosa.49 One of Wernicke’s three patients, in his original description, had had intractable vomiting as a result of pyloric stenosis induced by sulphuric acid poisoning.50
The classic features of the condition include an altered mental state, often with a disturbance of memory, coupled with a particular combination of neurological findings. Sometimes the mental state is normal, in other cases the patient presents in coma. Oculomotor signs are almost inevitable, typically nystagmus coupled with a lateral rectus palsy or gaze paresis. Vertical nystagmus is rather less common than horizontal nystagmus, but still occurs in about half of the cases. Gait ataxia is more common than limb ataxia. Most patients have evidence of a peripheral neuropathy. A failure to recognise atypical cases, without all the classic features, undoubtedly leads to underdiagnosis of the condition. In a postmortem series of 28 cases, only four had presented with the classic triad of altered mental state, ophthalmoplegia, and ataxia.51 Most of the patients in the series presented with an acute or chronic organic mental syndrome. Only nine of the 28 were recorded as having abnormal extraocular movements and/or nystagmus. The degree of recovery in patients with Wernicke-Korsakoff syndrome due to alcoholism is largely determined by the duration of history before admission and the delay in initiating thiamine therapy. Vertical nystagmus and any ophthalmoplegia recover completely, but horizontal nystagmus and gait ataxia often persist, and a transition from a global confusional state to a Korsakoff psychosis is commonplace.
NICOTINAMIDE DEFICIENCY
Endemic pellagra, linked to dietary deficiency of nicotinamide and associated with a triad of dermatitis, diarrhoea, and dementia, is no longer seen in developed countries. Nicotinamide deficiency has been described in some disorders of the alimentary tract. Bacterial colonisation of the small intestine can lead to the conversion of dietary tryptophan to indoles. In a patient with jejunal diverticulosis associated with bacterial overgrowth, a clinical syndrome of stupor, neck stiffness, rigidity, and grasp reflexes showed a dramatic response to nicotinamide.52 Non-endemic pellagra tends to lack dermatitis and diarrhoea. Its features then are similar to those encountered in alcoholic pellagra. Typically, patients are confused or display clouding of consciousness. Myoclonus is common, principally involving the face and shoulders. The third main element of the encephalopathy is Gegenhalten, which tends to spare the neck and predominate in the limbs.53 Less common signs include ataxia, pyramidal or cerebellar signs, primitive reflexes, seizures, and cranial or peripheral neuropathies.
VITAMIN D DEFICIENCY
Muscle weakness has long been recognised to occur in patients with metabolic bone disease. In an early report,54 a patient with steatorrhoea associated with jejunal villous atrophy presented with a year’s history of difficulty in walking. Examination disclosed a proximal weakness of both upper and lower limbs associated with a myopathic gait. Metabolic studies indicated osteomalacia. On a combination of a gluten free diet and vitamin D supplementation her motor deficit showed substantial improvement. In an analysis of 45 patients with osteomalacia with proximal myopathy, there were 14 with gluten enteropathy, five with a previous gastrectomy, and one with a history of distal small bowel resection.55 Almost inevitably the symptoms began in the lower limbs although examination often disclosed weakness of shoulder abduction and external rotation with weakness of elbow extension even in the absence of upper limb symptoms. In the legs, the weakness predominated in extension, flexion, and abduction of the hips. Peripheral muscles, at least in cases secondary to vitamin D malabsorption, were spared with intact reflexes and preserved sensation. EMG shows myopathic features with short duration polyphasic potentials. Changes on light microscopy of the affected muscles are not conspicuous. This and later studies have failed to show any correlation with the development of muscle weakness and the plasma calcium concentration. Indeed the exact mechanism of the muscle weakness remains unknown. Generally the muscle syndrome responds favourably to vitamin D.
VITAMIN E DEFICIENCY
Vitamin E deficiency may result from chronic fat malabsorption, as a part of cholestatic liver disease, in association with abetalipoproteinaemia, or as a familial disorder of vitamin E absorption.56 In patients with peripheral neuropathy secondary to vitamin E deficiency, peripheral nerve tocopherol (vitamin E) concentrations are depressed, a finding which antedates histological evidence of axonal degeneration.57 Abetalipoproteinaemia is an inborn error of lipoprotein metabolism leading to absence of apoprotein B. Neurological manifestations include neuropathy, cerebellar ataxia, ophthalmoplegia, and muscle weakness.58The absence of all the low density lipoproteins, consequent to the absence of apoprotein B, results in failure of absorption of vitamin E, concentrations of which are undetectable from birth in the serum of patients with the condition.59 A spinocerebellar disorder occurs in patients with cystic fibrosis or multiple ileal resections associated with undetectable serum concentrations of vitamin E and shows some response to vitamin E replacement therapy.60
A familial disorder of vitamin E deficiency has been described in the absence of any other gastrointestinal disturbance or lipid malabsorption.61 Patients absorb vitamin E normally but poorly conserve plasma α-tocopherol in very low density lipoproteins.56 The syndrome that emerges bears a striking resemblance to Friedreich’s ataxia (table 2).62 The patients have ataxia, cerebellar signs, dysarthria, bilateral extensor plantar responses, pes cavus, and scoliosis. Deep tendon reflexes are absent in the lower limbs, where there is impaired proprioception. Some of the patients have a cardiomyopathy. Genetic studies indicate that the disease is inherited as a recessive, the locus for which is not in the chromosome 9 region containing the gene for Friedreich’s ataxia, but rather, located on chromosome 8. Because of this overlap, all patients presenting with a clinical syndrome suggesting Friedreich’s ataxia should have vitamin E concentrations measured.
Clinical features of familial vitamin E deficiency
Coeliac disease
Coeliac disease is an intestinal disorder characterised by malabsorption, abnormal small bowel mucosa, and intolerance to the wheat protein gluten.63 Neurological symptoms are rare in children with coeliac disease. In adult series, however, neurological manifestations have been recorded in as many as 36% of patients,64 although figures of this magnitude have included cases of osteomalacic myopathy, cases of peripheral neuropathy or spinal cord disease secondary to vitamin B12 deficiency, and patients with episodic neurological dysfunction secondary to hypokalaemia or hypocalcaemia.
Neurological complications of less certain aetiology are recorded. A peripheral neuropathy, unrelated to B12 deficiency, produces predominant lower limb symptomatology with prominent ataxia, although some of this includes a cerebellar component. The neuropathy is usually progressive.65 Postmortem examination, in addition to showing evidence of peripheral nerve damage, has demonstrated cerebellar and spinal cord pathology, the latter with some of the features of subacute combined degeneration of the spinal cord. In general, the neuropathy is not influenced by the use of a gluten free diet. A detailed pathological study in a patient with coeliac disease who died after a progressive neurological disorder resistant to all appropriate nutritional measures has been published.63The neurological features included a neuropathy, cerebellar ataxia, and dementia. Eventually diffuse myoclonus emerged. Although the ankle reflexes were diminished, the others were exaggerated. Comparing this with previous studies, a pattern of neuropathological change emerges in which maximal damage is found in the cerebellum, brain stem nuclei and deep grey matter, and the spinal cord. Cerebellar findings are principally those of Purkinje cell loss with associated gliosis and loss of granule cells. Deep grey matter structures affected include the thalamus, caudate nucleus, globus pallidus, putamen, amygdala, anterior hypothalamic nuclei, periaqueductal grey matter, corpora quadrigemina, the substantia nigra, and the red nuclei. Various cranial nerve nuclei are affected. In the spinal cord, the changes concentrate in the posterior columns (mainly the fasciculi graciles) and the lateral columns. Cerebral cortical changes include focal neuronal atrophy and chromatolysis. The cause of this degenerative process has not been established. The pattern differs from the changes seen in alcoholic cerebellar degeneration and subacute combined degeneration or pellagra, and is uninfluenced by nutritional therapy.
In some patients with coeliac disease who develop progressive neurological dysfunction, the cerebellum bears the brunt of the process.66 67 Imaging in such patients has shown diffuse cerebellar atrophy.67 There are usually some, although inconspicuous, signs of neurological disturbance outside the cerebellum. As with cases with a more global encephalopathic syndrome, neuropathological studies have shown profound Purkinje cell loss to be the most consistent finding. In cases with adult coeliac disease with a spinocerebellar pattern of degeneration, no evidence of vitamin E deficiency has been detected as a possible explanation for the neurological process.68
In an early study, episodes of unexplained loss of consciousness were encountered in five of 16 patients with adult coeliac disease who had developed a progressive neurological disorder.65Subsequently a prevalence of epilepsy of 5.5% was recorded in a cohort of patients with coeliac disease, most having partial seizures.69 Some studies have confirmed the association and explored it in more detail. In one, 43 patients were selected either because of the unexplained association of cerebral calcification with epilepsy (31 cases) or because of the existence of epilepsy in established coeliac disease (12 cases).70 Among the first cohort of 31 patients, 24 were found to have unequivocal evidence of coeliac disease based on a full gastrointestinal evaluation including biopsy of the small intestine. Among the second group of 12 cases of coeliac disease with epilepsy, five were shown to have cerebral calcification on CT. The cerebral calcification was usually bilateral and predominated in the parieto-occipital regions (fig 3) Epilepsy, in the previously known cases of coeliac disease, had antedated the gastrointestinal disorder. In the whole group, the seizures were usually partial and predominantly occipital. There were no neurological signs in these cases. Some of the cases of epilepsy had proved to be drug resistant but tended to show an improvement once a gluten free diet was introduced in the previously undiagnosed cases. Alhough comparison has been made with the Sturge-Weber syndrome, significant differences exist. There is no evidence of enlargement of the choroid plexus and no evidence of cerebral atrophy. There is no evidence of abnormal deep cerebral veins, the calcification is usually bilateral rather than unilateral and neurological deficit has been found in a minority.71 Even in patients with a history of seizures and bilateral parieto-occipital calcifications but without abnormal intestinal biopsy, a diagnosis of coeliac disease may still be possible, based on the fact that the mucosal lesions in such cases may be patchy or of late onset, and on evidence of low folate concentrations of unknown cause in some of them.70 The aetiology of the cerebral changes has not been established.

{kind=link}
{kind=link}
{kind=link}
CT demonstrating bilateral parieto-occipital infarction in a patient with coeliac disease. (This figure was provided by Dr G Gobbi, Servizio di Neuropsichiatria Infantile Reggio Emilia, Italy.)
Inflammatory bowel disease
Thromboembolic complications are recognised features of both ulcerative colitis and Crohn’s disease. In some such cases, neurological involvement may follow. Cases of cerebral venous thrombosis are described in patients with ulcerative colitis.72 In one postmortem study of ulcerative colitis, thromboembolic disease was found in 39% of patients, predominating in the viscera and in the lungs.73 A similar predisposition to thromboembolic disease has been reported in patients with Crohn’s disease.74 In one case both arterial and venous thrombosis occurred, resulting in spinal cord ischaemia, although this case was complicated by the concomitant presence of hyperhomocysteinaemia.75 Cerebral arterial occlusions have been described affecting the internal carotid and middle and posterior cerebral vessels.76 Various factors have been suggested as the underlying trigger for the thromboembolism including, in the case of Crohn’s disease, focal induction of tissue factor procoagulant activity on endothelial cells and macrophages.
Peripheral neuropathy is a recognised complication of Crohn’s disease. In two cases, a sensory axonal polyneuropathy occurred, the severity of which waxed and waned according to the severity of the bowel disease.77 Neuropathy is more commonly encountered in patients with ulcerative colitis, and usually presents in the form of an acute or chronic inflammatory demyelinating polyneuropathy.78
Rarely, inflammatory bowel disease has been associated with muscle disorders. The underlying bowel disorder has usually been Crohn’s disease rather than ulcerative colitis. In one such case, a patient with a 12 year history of Crohn’s disease, requiring previous bowel surgery, developed typical clinical and investigative features of dermatomyositis.79 The condition responded to a combination of prednisone and azathioprine. In the other cases reported, the muscle involvement has ranged from non-specific changes on muscle biopsy to a florid picture of myositis with lymphocytic infiltration and muscle cell necrosis. Granuloma formation has been reported. It has been suggested, at least for cases with dermatomyositis, that immune complex formation, a known feature of inflammatory bowel disease, might be the mechanism for the muscle changes.
Neurological complications of inflammatory bowel disease may extend to the CNS. In a prospective study, brain MRI was performed in patients with Crohn’s disease or ulcerative colitis, and the findings compared with age matched controls. All the patients were under the age of 40. Hyperintense focal white matter lesions on T2 weighted images were found in 40%-50% of the patients with inflammatory bowel disease, but in only 16% of the control subjects.80 In most cases the lesions were single, and none enhanced. The distribution was not stated. None of the patients had neurological symptoms but the findings on neurological examination were not given. Although the authors reported either cardiolipin IgG or IgM antibodies in seven of the 72 patients with inflammatory bowel disease, the incidence in the control population was not stated. At present the pathogenesis of these changes, or indeed their confirmation in other series, has not been established. Myelopathy has been described with Crohn’s disease and, less commonly, with ulcerative colitis.
Whipple’s disease
Whipple’s disease was first described 90 years ago.81 Whipple noted the presence of rod shaped structures in vacuoles and even cultured a bacillus which he thought might be the responsible agent. Subsequently, light and electron microscopy studies confirmed the presence of rod shaped bacilli in the affected tissues, both, in the case of the bowel, lying free in the lamina propria of the small intestine and also as partly degraded structures within macrophage vacuoles.82 Subsequently, the same structures were located in the CNS, the heart, synovium, lymph nodes, lung, and liver. The demonstration of a distinctive pattern of bacterial antigens in the foam cells of affected patients served to confirm that only a single bacterium was concerned in the pathogenesis of the disease.83 Using nucleotide sequencing, it proved possible to demonstrate that the organism was a gram positive actinomycete, and a provisional title of Tropheryma whippeliihas been given to it.84 More recently the organism has been successfully propagated in cell culture although that propagation depended on the presence of interleukin-4.85
Some of the epidemiological aspects of the disease remain unexplained. It is rare, and is largely confined to North America and Europe. No specific immune defect has been detected in affected people. On the contrary, it has been suggested that the causative agent subverts the immune system by promoting interleukin-4 release or by blocking macrophage activating cytokines.82
Typically, the clinical manifestations of Whipple’s disease include diarrhoea, abdominal pain, weight loss, and joint pain. The disease presents in middle age.86 Evidence of CNS involvement was first established with the finding of PAS positive material in ventricular ependymal nodules and in perivascular accumulations.87 Subsequently, cases with clinical manifestations of the neurological pathology were published in which dementia and oculomotor signs were prominent features. In one such case, in which the diagnosis was established by jejunal biopsy, the patient became demented and complained of blurred vision with diplopia. His ocular examination disclosed fixed pupils, minimal nystagmus, and a fluctuating gaze paresis.88 The first report of a patient with Whipple’s disease confined to the CNS was published in 1977.89 The diagnosis was established at postmortem examination. The patient had presented with headache and focal seizures. Investigations established the presence of a left anterior temporal lobe mass which was later resected. Subsequently he became mute with primitive reflexes, Gegenhalten, and extensor plantar responses. Later he developed nystagmus although no other eye signs were recorded. Cases with more overt ophthalmoplegias have been presented.90 In one, a patient with drowsiness, impaired memory, and ataxia was found to be disorientated and lacking short term memory. Examination of extraocular movements disclosed a defect of saccadic upgaze with preserved pursuit. There was limb and truncal ataxia, and bilateral extensor plantar responses. Subsequently paralysis of upgaze emerged with absent doll’s head and caloric induced movements. Experience of these and other cases has established a particular pattern of CNS involvement. The findings, in decreasing order of frequency, include dementia, ophthalmoplegia (typically, at least initially, supranuclear in type), myoclonus, and various hypothalamic features including insomnia, hyperphagia, and polydipsia.91 92 The combination of convergence nystagmus with palatal, tongue, and mandibular movements called oculomasticatory myorhythmia has been considered pathognomonic. The parts of the brain particularly affected include the hypothalamus, cingulate gyrus, basal ganglia, insular cortex, and cerebellum.
Not surprisingly, diagnosis is difficult if the pathognomonic features are lacking. Magnetic resonance imaging identifies areas of reduced intensity on T1 and increased intensity on T2 weighted images which correspond with the known sites of pathological involvement.90 93 Gadolinium enhancement of the lesions has been described as has evidence of ependymitis. The CSF typically shows an inflammatory cell response, often including polymorphonuclear leucocytes, and sometimes contains PAS positive macrophages. Chloramphenicol and trimethoprim-sulphamethoxazole are capable of arresting the course of CNS disease with gaze palsies and nystagmus being the most responsive signs.
Summary
Both achalasia and Hirchsprung’s disease arise from defects of innervation of the oesophagus and distal large bowel respectively. Their consequences are confined to disorders of motility in the relevant part of the gastrointestinal tract. Many neurogenic and primary muscle disorders are associated with abnormalities of gut motility. Stroke, even when unilateral, is commonly associated with dysphagia. Transcranial magnetoelectric stimulation has established that the pharyngeal phase of swallowing tends to receive its innervation principally from one hemisphere. In many neurological disorders, dysphagia is only one part of the clinical picture but in some—for example, the Chiari malformation—dysphagia may be the sole or major feature. Disturbances of small and large bowel motility, when seen in neurogenic disorders, are associated with autonomic neuropathy and are particularly common in diabetes mellitus. Primary muscle disorders can lead to dysphagia (for example, with polymyositis or oculopharyngeal dystrophy) or defects of large bowel motility (for example, with Duchenne’s muscular dystrophy). Primary gut disorders particularly associated with neurological disease include pernicious anaemia, nicotinamide and thiamine deficiencies, selective vitamin E deficiency, and coeliac disease. Inflammatory bowel disease is associated with thromboembolic complications which may include the CNS, inflammatory muscle disease, and abnormalities on MRI of the brain of uncertain relevance. Whipple’s disease is a rare condition which sometimes is largely or entirely confined to the CNS. In such cases, a particular neurological presentation can indicate the diagnosis.