Article Text

Abstract
Background Dementia is a common, debilitating feature of late Parkinson’s disease (PD). PD dementia (PDD) is associated with α-synuclein propagation, but coexistent Alzheimer’s disease (AD) pathology may coexist. Other pathologies (cerebrovascular, transactive response DNA-binding protein 43 (TDP-43)) may also influence cognition. We aimed to describe the neuropathology underlying dementia in PD.
Methods Systematic review of autopsy studies published in English involving PD cases with dementia. Comparison groups included PD without dementia, AD, dementia with Lewy bodies (DLB) and healthy controls.
Results 44 reports involving 2002 cases, 57.2% with dementia, met inclusion criteria. While limbic and neocortical α-synuclein pathology had the strongest association with dementia, between a fifth and a third of all PD cases in the largest studies had comorbid AD. In PD cases with dementia, tau pathology was moderate or severe in around a third, and amyloid-β pathology was moderate or severe in over half. Amyloid-β was associated with a more rapid cognitive decline and earlier mortality, and in the striatum, distinguished PDD from DLB. Positive correlations between multiple measures of α-synuclein, tau and amyloid-β were found. Cerebrovascular and TDP-43 pathologies did not generally contribute to dementia in PD. TDP-43 and amyloid angiopathy correlated with coexistent Alzheimer pathology.
Conclusions While significant α-synuclein pathology is the main substrate of dementia in PD, coexistent pathologies are common. In particular, tau and amyloid-β pathologies independently contribute to the development and pattern of cognitive decline in PD. Their presence should be assessed in future clinical trials where dementia is a key outcome measure.
Trial registration number CRD42018088691.
- Parkinson's disease
- dementia
- neuropathology
Statistics from Altmetric.com
Introduction
Cognitive impairment, including dementia, is a common and debilitating feature in patients with Parkinson’s disease (PD). PD dementia (PDD) is distinguished from dementia with Lewy bodies (DLB) by the relative timing of the onset of cognitive symptoms versus motor signs. In PDD, the onset of dementia is usually several years after PD diagnosis; in DLB, cognitive symptoms occur within 1 year of the diagnosis of parkinsonism, or precedes it.1 2 However, patients with PD may have other causes of cognitive impairment and dementia, such as comorbid Alzheimer’s disease (AD) or cerebrovascular disease. These pathologies may be difficult to identify in vivo, as the clinical features often overlap with those of PDD, and there are no definitive biomarkers. The differentiation of dementia type is an important consideration in clinical research, as treatments under development are specifically targeted against abnormal accumulation of α-synuclein, which is the pathological hallmark of PDD and DLB,3 or tau or amyloid-β, which underlie AD.4 5 A better understanding of the contribution of different pathological substrates to the development of dementia in PD would be useful for the planning and analysis of the efficacy of these new treatments.
Autopsy studies are the definitive approach to identify the pathological basis of dementia in PD. They also ensure that the clinical diagnosis of PD is accurate, which is important given known diagnostic error rates in vivo.6 7 We conducted a systematic review of autopsy studies in PD cases with dementia, to define the relative role of α-synuclein, tau, amyloid-β and other pathologies, including transactive response DNA-binding protein 43 (TDP-43) and cerebrovascular disease, in the aetiology of cognitive decline in PD.
PD is pathologically defined by two key features. Degeneration of the nigrostriatal dopaminergic system underlies the progressive motor syndrome that clinically defines the disease.8 Accumulation of misfolded, insoluble aggregates of α-synuclein protein in the neuronal perikarya (Lewy bodies) and processes (Lewy neurites) is the second hallmark. According to the Braak staging model for PD (Braak-PD staging), α-synuclein pathology spreads in a stereotypical, caudal-to-rostral pattern in PD, from the brainstem through the limbic system to the neocortex. Propagation of α-synuclein aggregates to the neocortical regions is the main substrate of cognitive decline in PDD and DLB.1 2
PDD is neuropathologically distinct from AD,9 the most common cause of dementia. Like PD, AD is defined at autopsy by two hallmark lesions. Neurofibrillary tangles are insoluble, intraneuronal bundles of hyperphosphorylated tau protein. Amyloid-β plaques are extraneuronal structures composed primarily of amyloid-β peptide. ‘Neuritic plaques’ are amyloid-β aggregates with dystrophic tau-positive neurites that are characteristic of AD.
Braak staging for tau pathology10 defines the spread of tau-positive tangles and neuropil threads from the transentorhinal cortex (stages I–II), into the entorhinal cortex and hippocampus (stages III and IV), and finally to neocortical areas. Amyloid-β pathology is described in five Thal phases,11 from the neocortex to the entorhinal cortex and subcortical nuclei, and finally to the brainstem and cerebellum. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) protocol12 is widely used to grade the severity of neuritic plaques.
In addition to Lewy and Alzheimer type abnormalities, various other pathologies can underlie dementia. TDP-43 aggregates contribute to cognitive decline in most forms of frontotemporal dementia,13 and cerebrovascular disease (infarcts, haemorrhages and white matter lesions) contributes to vascular dementia. The specific pathology underlying vascular dementia varies, but primary causes are cerebral amyloid angiopathy (CAA), atherosclerosis, and cerebral small vessel disease.14
This novel systematic review included autopsy studies of pathologically verified PD cases with a history of dementia. The primary objective was to define the extent of Lewy, Alzheimer and other pathologies (cerebrovascular, TDP-43) in cases of PD with dementia.
Methods
This review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses.15 The protocol was predefined and registered with the Centre for Reviews and Dissemination’s International Prospective Register of Systematic Reviews at www.crd.york.ac.uk/prospero/ on 13/02/2018.
Autopsy studies of pathologically verified PD, including cases with dementia diagnosed prospectively or retrospectively, were identified. The minimum sample size was five (including cases and controls). Only full text papers in English presenting original data were included.
Comparators or controls were not mandatory for inclusion, but many studies compared PD cases with dementia to other groups, including PD cases without dementia, DLB, and AD.
The primary outcome was the presence of dementia during life, defined by clinical diagnosis or by at least one objective test of cognitive function. Studies limited to PD with mild cognitive impairment were not included.
Search criteria were piloted in five key articles, and then applied from 1946 (Medline, Ovid) or 1974 (Embase, Ovid) to January 2018. Medical subject headings (Parkinson/disease/pa, Dement*.mp, Cogniti*.mp) were combined and filtered to humans. Duplicates were removed. Search terms and syntax were adapted for LILACS (BIREME), CINAHL (EBSCO) and the Cochrane Library (Wiley), all up to January 2018. Reference lists within included articles and relevant reviews were handsearched, and the ‘cited by’ function was applied to all records ultimately included in the review. The search terms were initially piloted for sensitivity by confirming that they identified five pre-selected key articles. The search strategies for each database are provided in the online supplementary material.
Supplemental material
Preliminary screening relied on title and/or abstract screening, then relevant articles were read in full. Both stages of the selection process were independently conducted by two authors (CRS and NMM), with a third (KAG) resolving discrepancies.
Data describing the type, location and extent of pathological findings (including staining methods and criteria) were extracted, and statistical comparison methods were noted. Studies were individually assessed by one author for risk of selection, detection and reporting bias, using a modified version of the semiquantitative Cochrane tool.16 The initial plan to use a modified Newcastle-Ottawa scale for this purpose was abandoned due to concerns about the scale’s validity.17 Eligible studies were included in the review regardless of risk of bias.
Some reports used the same cases; in determining the sample size of all studies combined, these cases were only counted once. Partial overlap between studies was not subjected to any adjustment because of the small numbers involved. In generating the bar charts to describe the severity of Alzheimer pathology, it was sometimes necessary to impute data where categories in the original report (eg, absent and mild) had been collapsed. Values for imputation were based on average values when all studies were combined. Aggregation was done for each bar chart based on raw data.
Results
Study selection and characteristics
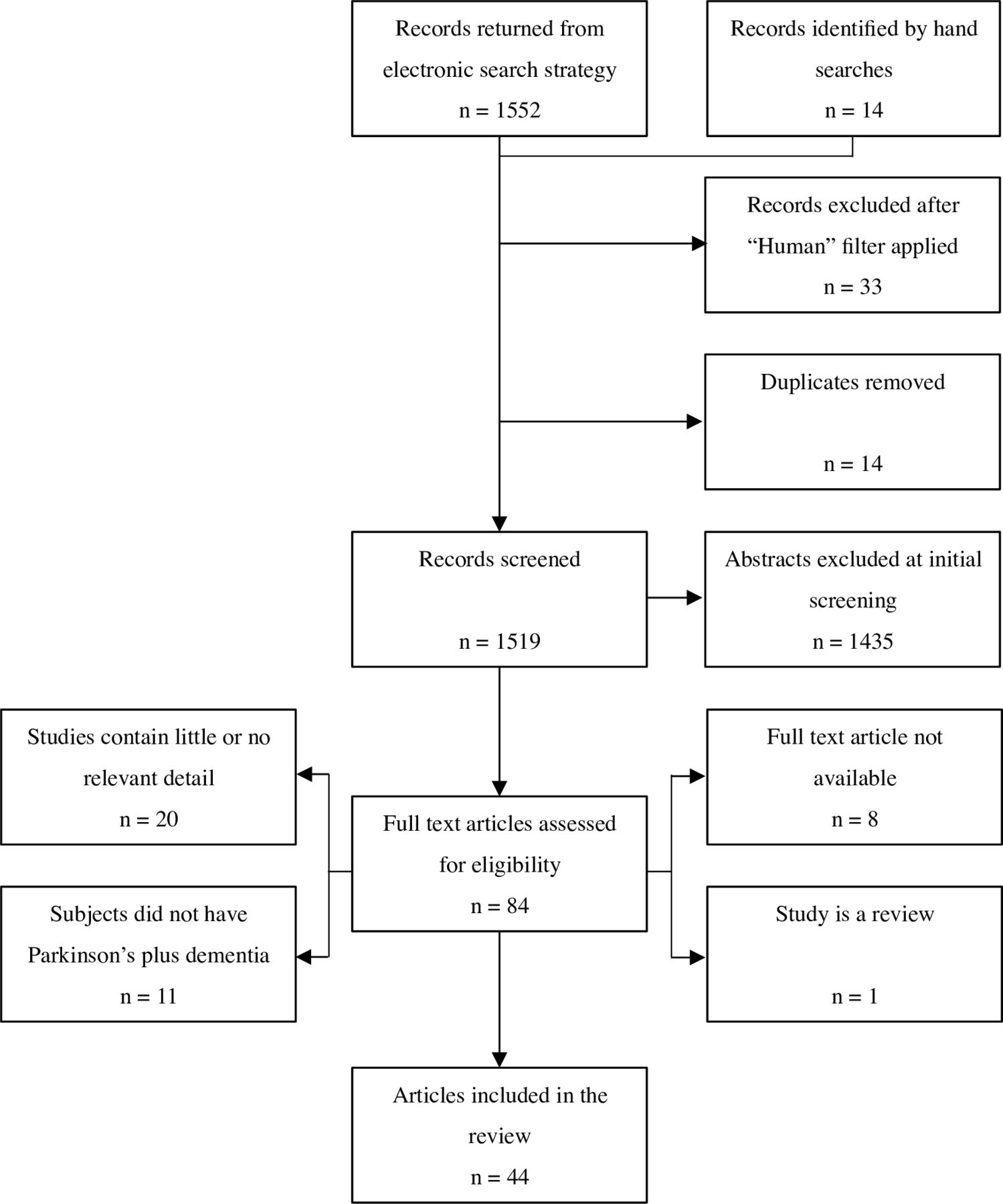
Electronic searching retrieved 1552 records, and 14 more were identified from hand searching. Excluding duplicates and non-human studies, 1519 records underwent preliminary screening; 84 were potentially relevant and were read in full. In all, 40 of these were discarded, in most cases due to the lack of relevant detail or PD cases with dementia. Ultimately, 44 articles involving 41 study groups were included (figure 1). Inter-rater agreement was 96.8% at initial screening, and 93.6% at the full-text stage.
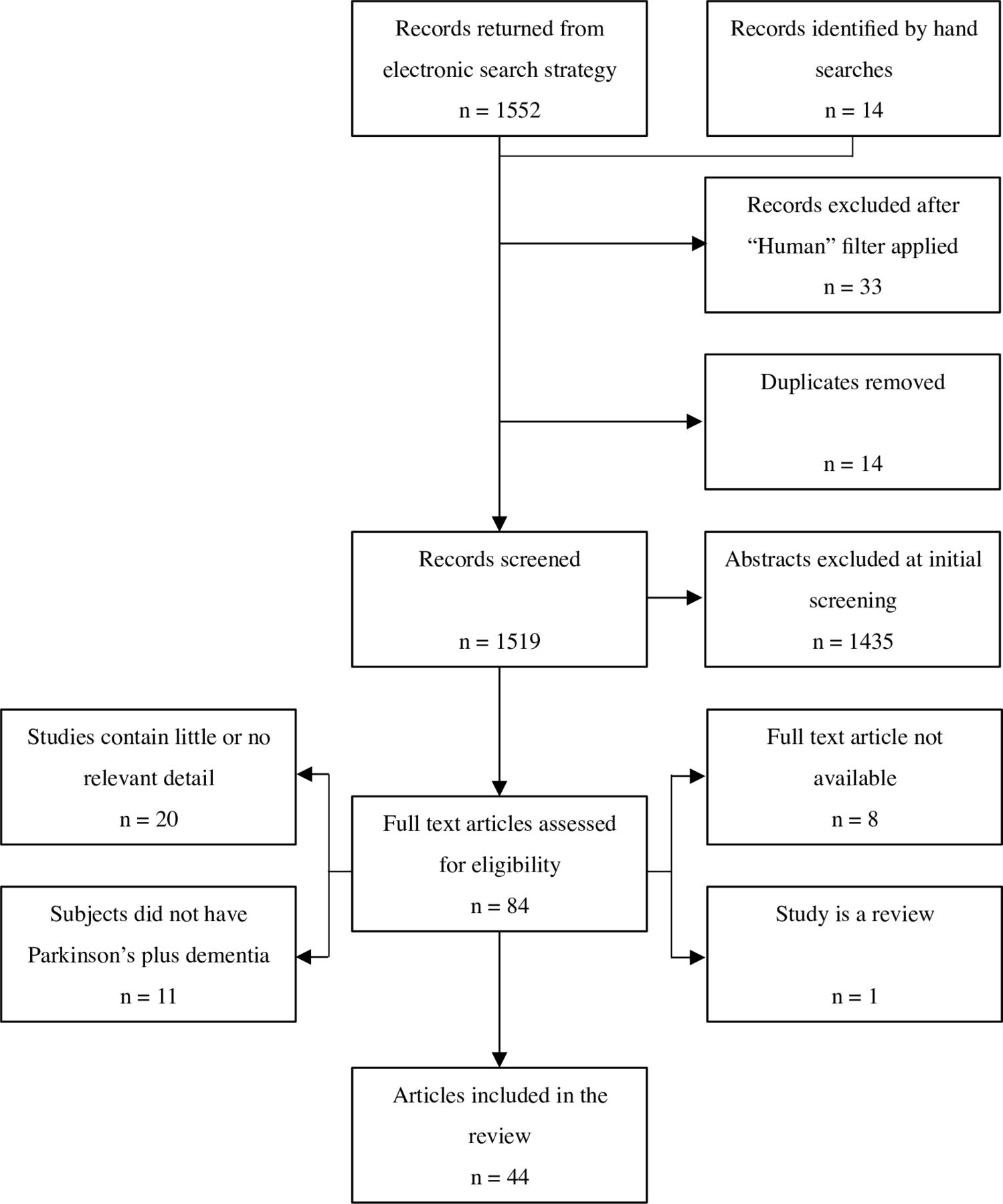
PRISMA flow diagram showing selection of autopsy studies for inclusion in the systematic review. PRISMA, Preferred Reporting Items for Meta-analyses and Systematic Reviews.
The 44 reports involved 2002 cases with clinically diagnosed and pathologically verified PD, 1145 (57.2%) of whom had dementia. Dementia was primarily a clinical diagnosis, generally referencing the Diagnostic and Statistical Manual of Mental Disorders, but standard cognitive tests were often applied. Comparison groups included PD without dementia, DLB, AD and healthy controls (table 1).18–61
Characteristics of reports included in the systematic review
Seven studies excluded cases with coexistent tau pathology,24 25 31 32 44 47 61 and one study required cases to have both Lewy and Alzheimer pathologies.59 Differential diagnosis of PDD versus DLB typically relied on the 1-year latency rule.1 2 However, some studies excluded cases with cognitive decline within 2–5 years of motor onset.19 32 36 37 47 57
Risk of bias
Overall, 77.3% of reports had a low risk of bias. Nine had an unclear risk, and one had a high risk of bias. Selection bias was unclear in 13 reports (29.5%) that did not report the source of autopsy cases. Two studies had a high risk of detection bias. The cases in one study were primarily hospital residents,48 and in the other, tissue samples were collected from National Institute on Ageing (NIA)-funded AD centres52; either procedure may have led to datasets that do not represent at typical PD populations. Detection bias was unclear in 24 (54.5%) reports due to omission of information about the pathological assessment procedures, including what stains were used, whether or not the neuropathologists were blind to clinical data and what guidelines or diagnostic criteria had been consulted. Reporting bias was high in one study48 that focused almost entirely on a single case in the results section, and omitted relevant medical and neuropsychological information for other cases. All other studies had a low risk of reporting bias (online supplementary table 1).
α-synuclein pathology
Lewy pathology was detected by routine staining or antibodies to ubiquitin in earlier studies while antibodies to α-synuclein were widely used following their introduction in 1998.62 Two early studies found leading causes of dementia in PD to be comorbid AD, cortical Lewy body deposition and cerebrovascular disease, though no cause was found in a significant percentage,35 42 probably due to older staining methods.
Dementia was linked to Braak-PD or McKeith stage in several studies in a total of 480 cases. In PD cases with dementia, α-synuclein pathology almost invariably extended to the limbic system or neocortex (85.3%–100% of cases); neocortical involvement was more frequent than in PD without dementia in all four studies that conducted a comparison.26 33 37 45 Insoluble cortical α-synuclein levels were particularly overexpressed, though soluble levels were also raised, according to an analysis of the inferior frontal gyrus.61 Limbic and neocortical Lewy body counts were approximately 10 times higher in dementia cases.19 A moderate total cortical Lewy body score had excellent sensitivity (90.9%) and specificity (90.0%) for detecting dementia in PD.36 The severity of cognitive impairment correlated strongly with Braak-PD stage,23 and with Lewy body densities in the frontal, straight, angular, cingulate and middle temporal gyri, particularly when cases with coexistent Alzheimer pathology were excluded.49 51
PD cases with dementia also showed higher Lewy pathology in subcortical regions relative to cognitively healthy PD cases. In the striatum, insoluble α-synuclein levels were twice as high.61 In the amygdala and hippocampus, studies relying on ubiquitin stains found no association of Lewy bodies to dementia,24 28 50 but studies using α-synuclein stains found higher Lewy body densities in these regions that correlated with dementia severity.19 31 51 Parahippocampal α-synuclein scores had excellent sensitivity (91%–93%) and specificity (84%–88%) for separating PD cases with and without dementia.32 However, in the nucleus basalis, substantia nigra and midbrain, no differences in α-synuclein burden were seen between PD without dementia, PDD and DLB.19 56
DLB groups tended towards greater global α-synuclein pathology than PDD, but differences were usually not significant.29 31 54 Striatal α-synuclein differentiated PDD (29.4% of 17 cases) from DLB (76.5% of 17 cases, p<0.001) in one study.40 Additionally, in the claustrum, α-synuclein deposition rose progressively from PD without dementia to PDD to DLB.43
The strong association between extensive α-synuclein pathology and dementia was challenged by some observations. Some large studies found that 15.2%–44.7% of 181 cognitively healthy PD cases had severe, neocortical-type pathology.26 33 37 45 One study described 17 PD cases with no dementia despite limbic and/or neocortical pathology, and concluded that no clear threshold of Lewy body burden can distinguish PD cases with and without dementia.25
The opposite phenomenon, dementia cases with modest α-synuclein pathology, was also reported. In one study, three PD cases with dementia had no Lewy bodies outside the brainstem, and only mild or absent Alzheimer or vascular pathology.48 Dementia cases with α-synuclein pathology confined to the brainstem were sporadically reported elsewhere: such cases accounted for 14.7% of one study of 109 dementia cases,33 but most studies reported much lower figures.18 23 25 26 32 37 46 54 56 59
Alzheimer pathology
Coexistent tau and amyloid-β pathology of varying severity was common in PD cases. Tau pathology was universally scored with Braak-tau staging. A third of cases with dementia had moderate or severe tau pathology (stages III and VI). There was heterogeneity in the methods used to quantify amyloid-β lesions. Most studies used CERAD criteria, and therefore considered neuritic plaque burden only. Moderate to severe amyloid-β pathology affected just over half of PD cases with dementia (figure 2).

{kind=link}
{kind=link}
Severity of coexistent Alzheimer pathology in cases of Parkinson’s disease with dementia. (A) Severity of amyloid-β pathology using CERAD criteria, except for Horvath et al, who used Thal phases, and Braak et al, who used a semiquantitative scoring method. (B) Severity of tau pathology by Braak-tau staging. (C) Likelihood of Alzheimer’s disease by NIA-Reagan criteria. aData were imputed for two or more categories. bThere was potential overlap between these cases. CERAD, Consortium to establish a Registry for Alzheimer’s disease; NIA, National Institute on Ageing.
The proportion of PD with comorbid AD varied markedly. The four largest studies (n=88 to n=200) that defined AD as intermediate or high probability by NIA-Reagan criteria (accounting for both plaques and tangles) were reasonably consistent: comorbid AD was diagnosed in 19.3%,23 20.0%,33 28.6%37 and 31.5%39 of PD cases. Three of these four studies reported the rate of comorbid AD by these criteria in PD cases with dementia, where there was more variation: 21.5%,23 38.2%,37 and 89.4%39 (figure 2).
Some early studies linked dementia in PD to marked depletion of cholinergic neurons in the basal forebrain and cortex,30 53 60 as also typifies AD. Comorbid AD was associated with greater cortical and amygdala atrophy in PD cases with dementia, but the striatum was severely atrophic regardless of comorbid AD.27
Tau and amyloid-β pathologies in PD cases with dementia were typically graded as moderate to severe only in the entorhinal cortex, and mild in the hippocampus, according to two descriptive studies. The neocortex was generally unaffected by tau and variably affected by amyloid-β.22 28 The prefrontal cortex was more affected by tau pathology than the temporal cortex in PDD cases (a rare pattern in AD); the occipital and cingulate cortices were rarely affected.58
Two reports based on the same study cohort described advanced Alzheimer pathology in PD cases with severe dementia, and concluded that dementia in PD was due to comorbid AD.21 38 Later studies from the same research group supported this.39 41 Alzheimer pathology was substantially higher in these studies than in other series (figure 2).
Striatal amyloid-β measures differentiated PDD from DLB. All of 17 DLB cases had moderate to severe striatal amyloid-β in one study, compared with only 17.6% of 17 PDD (p<0.001).40 A study excluding cases with significant neurofibrillary pathology similarly found higher striatal amyloid-β in DLB, which correlated with dementia severity, and had 100% sensitivity and 66.7% specificity for differentiating the 10 DLB cases from 12 PDD cases.31 In another study, 92.9% of 14 DLB cases had dense-core plaques in the striatum, which were entirely absent in 41 PDD cases. The higher frequency of severe striatal amyloid-β in DLB was not significant in this analysis.44
Midbrain and cerebellar amyloid-β was higher in DLB than PDD in one study of 30 combined cases,29 but not in another of 20 cases, possibly because that study involved PDD cases that were approximately 7 years younger on average.56 In both studies, subtentorial tau was comparable in PD (regardless of dementia) and DLB. Claustrum amyloid-β was comparable between PDD and DLB, but exceeded levels in cognitively healthy PD, while tau was negligible in all three groups.43 Finally, in cases with comorbid Lewy and Alzheimer pathologies, greater global amyloid-β (significant in the temporal and cingulate cortices) and tau (significant in the frontal and cingulate cortices) was found in those diagnosed clinically with DLB, relative to PDD.59
TDP-43, argyrophilic grain and cerebrovascular pathologies
Only one study examined TDP-43 pathology in PD cases with comparators.52 Hippocampal and entorhinal slides were positive in 7.2% of 69 PD without dementia, 19.0% of 21 PDD, 31.3% of 80 DLB plus AD, none of the 10 DLB cases without AD, and 3% of 33 healthy controls. Significant differences were found only when comparing a disease group to healthy controls, and a dementia group with a non-dementia group.
Two studies reported argyrophilic grain disease in approximately 5% of PD cases (n=88 and n=140), with no association with dementia.23 37
Cerebrovascular pathology was found in 16.7%–28.6% of cognitively healthy PD cases, and 15.6%–44.4% of those with dementia,18 33 37 54 though the definition of vascular pathology varied between studies. Individually, none of these studies found a significant association between cerebrovascular disease and dementia. Combining the results of the individual studies did not produce a significant result (total n=421, χ 2=0.01, p=0.92).
Unlike other types of cerebrovascular pathology, CAA was significantly more common in dementia.26 37 Capillary CAA was most often severe in PD cases with dementia (>50%); generalised CAA affecting meningeal and cortical vessels was severe in around a quarter, and moderate in around a third. In contrast, over three-quarters of cognitively healthy PD cases were completely unaffected.41
Relative contribution of α-synuclein, amyloid-β, and tau to dementia
Multiple indices of abnormal α-synuclein, amyloid-β and (less consistently) tau were significant predictors of dementia in PD based on univariable regressions.33 37 54 One study of 22 PD cases found that most of the variance in cognitive scores was accounted for by Lewy pathology in the entorhinal, anterior cingulate and temporal cortices, with smaller contributions from entorhinal and temporal amyloid-β.47 The contribution of amyloid-β and tau was typically lower by multivariable regression (table 2). Braak-tau stage remained independently associated in one study33 but Lewy pathology was consistently the best predictor. The predictive values of amyloid-β and tau were lost in another model when cortical Lewy bodies were added; strong collinearity of these pathologies precluded further multivariable analysis.26
Significant predictors of dementia in Parkinson’s disease by multivariable regression
One study of 104 cases used receiver operating characteristic curves to predict dementia in PD, and found that mean cortical Lewy body score alone was the best predictor (area under the curve (AUC) 0.80, 95% CI 0.72–0.88).54 However, another study of 56 cases indicated that diagnostic accuracy was improved by the addition of indicators of amyloid-β and tau pathology (AUC 0.95, 95% CI 0.88 to 1.00).26
A multivariable regression examining dementia severity found that anterior cingulate and entorhinal Lewy body densities together accounted for approximately 60% of the variance in cognitive scores; values for amyloid-β and tau were not significant.47 Linear regression in another study again found the best fit for cingulate Lewy pathology as a predictor of dementia severity; additionally, temporal neurofibrillary tangle load was a marginally significant predictor. When cases with severe amyloid-β load were excluded, frontal α-synuclein was the sole significant predictor.51
Only one study (n=22) found that cognitive scores were unrelated to any measure of α-synuclein, amyloid-β or tau pathology, though the Lewy body score predicted the annual rate of cognitive decline.18 The best predictor of annual decline was a summated score incorporating both Lewy and Alzheimer pathologies of the prefrontal cortex, according to a study that used multiple backward regressions.34
None of the studies that assessed the value of cerebrovascular disease pathology (including CAA) for predicting dementia found a significant association, using univariable or multivariable regression methods.26 33 37 54
The motor–cognitive interval and mortality
The latency from motor to cognitive onset in PD was studied in four survival analyses20 46 54 55 with a combined total of 166 PD cases of dementia; three of these studies20 54 55 associated higher amyloid-β with a shorter motor–cognitive interval. Moderate to severe plaque pathology reduced the motor–cognitive interval by 4.7 years on average.55 Additionally, a multivariable regression model established cortical amyloid-β as the only significant predictor of a shorter motor–cognitive interval.54 No associations of α-synuclein or tau pathologies with the motor–cognitive interval were found in any of these studies.
In another study (n=28), a longer time to dementia correlated moderately with lower plaque scores, and more weakly with less severe α-synuclein pathology, but not with Braak-tau stage. A significant correlation (r=–0.37, p=0.04) was also observed with the amount of choline acetyltransferase in the temporal cortex, indicating that cholinergic activity in this area is reduced in cases with a longer duration of PD preceding cognitive onset.20
Other studies explored the association of pathology with mortality. One study (n=32) linked increased amyloid-β pathology to higher mortality using Kaplan-Meier curves.46 A larger study (n=200) additionally found a weaker association of tau pathology to mortality. Coexistent Alzheimer lesions decreased life expectancy by a mean of 4.5 years. Multivariable regression found that dementia, tau pathology, and particularly, neuritic plaque pathology all predicted lower survival.39
Interaction between neuropathological disease processes
Moderate correlations were reported between Braak-tau stage and cortical and striatal amyloid-β load26 31 and Thal phase.33 Alzheimer lesions also correlated positively with α-synuclein burden. Braak-tau stage correlated weakly with Braak-PD stage, global Lewy body score, and global, cortical, and striatal Lewy neurite scores.26 31 33 Stronger correlations were generally found between α-synuclein and amyloid-β. Global Lewy body scores correlated better with Thal phase than Braak-tau stage, though the coefficient was still modest.33 Moderate correlations were found between multiple indices of α-synuclein pathology (Braak-PD stage, cortical Lewy body score and striatal and hippocampal Lewy neurites) and cortical and striatal amyloid-β pathology.26 31 Another study demonstrated moderate correlations between striatal α-synuclein and cortical and hippocampal plaques, but not with hippocampal tangles.57
CAA correlated moderately with tau pathology, and more modestly with cortical amyloid-β load.26 Similarly, TDP-43 pathology correlated strongly with Braak-tau stage52; unfortunately, the potential association with amyloid-β was not analysed.
Discussion
The results of the systematic review indicate that limbic and neocortical α-synuclein pathology is present in virtually all cases of PD with dementia. Coexistent pathologies that contribute to dementia, most notably Alzheimer-related changes, are common. Of the studies that defined pathological AD by NIA-Reagan criteria, between a fifth and a third of all PD cases fulfilled that diagnosis.23 33 37 39 In the PD cases with dementia, tau pathology was moderate or severe in around a third, and amyloid-β pathology was moderate or severe in just over half. Even taking account of differences across studies, we can reliably conclude that tau and amyloid-β pathologies are both common in PD cases at autopsy, particularly those with dementia.
The relative contribution of Alzheimer versus α-synuclein pathologies was more difficult to define, due to variation between studies. Reports from one research group, with some overlap in study samples, found advanced Alzheimer pathology in most PD cases with dementia.21 38 39 41 In other studies, Alzheimer pathology was universally less frequent and less severe. These differences were not explained by systemic differences in dementia severity, age, disease duration or case selection procedures. While global tau indices independently predicted dementia in PD cases in one study,33 two other studies found no such association.37 54 One possible explanation for this was the lower α-synuclein burden in the first study compared with the other two, which might have allowed the independent contribution of tau to emerge. Another factor may be the distribution of tau pathology; neurofibrillary tangles in the temporal lobe were linearly associated with dementia severity in another study.51
Combined, these results indicate that tau pathology contributes to dementia in a subset of PD cases, particularly when α-synuclein deposition is relatively low or tau deposition relatively high. Amyloid-β, in contrast, was not independently related to dementia in any study. Thus, tau has a closer relationship with cognitive status in PD than amyloid-β, which is consistent with observations in AD.63 These findings have implications for clinical trials of disease-modifying immunotherapies in PD: using biomarkers to quantify coexistent tau pathology will be helpful, particularly for studies that include cognitive impairment as a key outcome. The value of immunotherapies targeting tau deposition in PD will need to be evaluated.
While amyloid-β pathology was not associated with the presence of clinical dementia diagnosis in PD, moderate to severe deposition was strongly linked to a more rapid cognitive deterioration,20 31 54 55 as well as earlier mortality.39 46 Longitudinal studies measuring amyloid-β in vivo (eg, by cerebrospinal fluid assays or neuroimaging) support its association with the rate of cognitive decline in PD, showing that higher amyloid-β predicts a greater decrease in cognitive scores.64 65 Identifying amyloid-β deposition is therefore likely to become relevant in disease-modifying PD trials, either as part of selection criteria or as a stratification factor for sub-group analyses.
The consistent association of amyloid-β with the motor–cognitive interval in both autopsy and biomarker studies indicates that it is a key factor distinguishing DLB from PDD. The most striking differences were in the striatum. Moderate–severe striatal amyloid-β had good specificity (66.7%–82.4%) for DLB against PDD,31 40 and dense-core striatal plaques were present only in DLB.44 Neuroimaging findings indicate that these differences are detectable in vivo, with DLB cases showing higher cortical, cingulate and striatal amyloid-β than PDD cases.66 It remains unclear how early these differences emerge; further studies of this would help to refine the diagnostic criteria for prodromal PD. These criteria rely on features such as rapid eye movement sleep behaviour disorder, hyposmia and autonomic dysfunction,67 all of which also frequently precede DLB, such that the criteria often capture cases who progress to DLB rather than PD.68 Amyloid-β biomarkers, which are already incorporated into criteria for prodromal and preclinical AD,69 70 may also have a place in prediction of PD versus DLB.
The relationship between α-synuclein deposition and dementia was strong, as expected, but there was again some variation between studies. Global cortical α-synuclein was generally the best predictor of dementia,33 37 47 51 54 though cingulate or frontal scores had the best correlation in one study,51 and the addition of tau and amyloid-β scores improved predictive accuracy for dementia in another.26 Neocortical, limbic and paralimbic α-synuclein burden was almost universally more severe in dementia cases and had excellent sensitivity and specificity for distinguishing these cases from PD without dementia.32 36 While these findings strongly support Braak theory71 and the role of extensive α-synuclein deposition as the primary substrate of dementia in PD, some exceptions to this rule were observed.
First, significant α-synuclein deposition in limbic or neocortical areas was often found in PD cases who did not have a history of cognitive impairment. Such cases accounted for around 15%–45% of the cognitively healthy PD group in the largest studies.26 33 37 45 Other large autopsy studies similarly reported relatively severe α-synuclein, as well as tau and amyloid-β pathologies, in many elderly cases with neither motor nor cognitive impairment during life.72 73 Higher cognitive reserve may explain the variance between pathological and clinical severity in AD,74 and this could also apply to PD.75 Unfortunately, too few studies reported proxy measures of cognitive reserve, such as educational or occupational attainment, to allow us to test this. Neural plasticity, genetics, and environmental exposures may also explain the variance in these observations; all of these are worthy of further study in the context of PD.
Second, a rare but intriguing finding was the occurrence of dementia cases with relatively modest, brainstem-type α-synuclein pathology. The cognitive decline in these cases is presumably due to a pathology distinct from the primary condition. Comorbid AD probably accounts for the majority of these, but some individual cases may have had other problems, including vascular or frontotemporal dementia. Alternatively, cholinergic dysfunction originating in the basal forebrain, which was related to cognition in several studies,30 53 60 might be sufficient to independently cause dementia in a small number of cases.
Several other pathologies may contribute to dementia in PD. CAA was significantly more common in dementia cases, and correlated with coexistent Alzheimer pathology.26 37 41 Other cerebrovascular pathologies were not more common in dementia,18 33 37 54 even when the studies were combined. TDP-43 did not contribute to dementia in the only study that assessed this; while it correlated strongly with tau pathology,52 the correlation with amyloid-β pathology was not assessed. Animal models indicate that amyloid-β induces TDP-43 misfolding and aggregation76; the potential for this in PD could be examined in future research. Argyrophilic grain disease was rare and not associated with dementia in PD,23 37 and is likely an incidental or age-related finding. Thus, while these molecular pathologies are common in PD—particularly in dementia cases, and most of all in cases with comorbid AD—they do not generally contribute to cognitive decline.
Finally, previous in vitro and animal studies indicate that α-synuclein, amyloid-β and tau may promote one another’s aggregation to accelerate neurodegeneration and dementia.77–81 Multiple studies in the present review found correlations that support this theory in PD cases.19 26 31 33 51 This suggests that disease-modifying therapies targeted against one pathology may inhibit the proliferation of another, increasing the overall benefit. This effect has been observed in a murine model, in which mice overexpressing α-synuclein were protected from both cognitive and motor impairment after treatment with an antibody for tau.82 How this would translate to clinical research involving humans remains to be evaluated.
Limitations
Methodological limitations in the studies assessed included small sample sizes in many, and an absence of power calculations. Dementia ascertainment was retrospective in 32 reports (72.7%), being derived from non-standardised medical notes. The retrospective distinction of PDD from DLB may be difficult,83 though some studies required a longer motor–cognitive interval to exclude ambiguous cases.19 32 36 37 47 57 Several studies assessed cognition objectively but used short screening instruments (eg, the Mini-Mental State Examination) with limited sensitivity and specificity for dementia in PD.84 Almost a third of studies did not report the source of the autopsy cases, and more than half omitted information about the neuropathological assessment procedures.
Finally, differing protocols for the pathological assessment of amyloid-β sometimes made comparisons difficult. Newer studies are likely to use the most recent NIA criteria for pathological AD,85 which incorporate both Thal and CERAD scoring; this will help overcome this variation in future research. In the present review, only a limited number of studies applied NIA-Reagan criteria,18 19 29 39 and there was major variation in the proportion of cases with comorbid AD between these studies.
The review itself was potentially limited by aspects of the electronic search strategy. There was some potential for publication bias: eight potentially relevant unpublished articles were found that could not be retrieved online. Moreover, the exclusion of articles in languages other than English (n=6, titles provided in online supplementary material) may have led to the omission of some relevant detail.
Conclusions
Several disease processes (particularly Alzheimer related) are common in PD and are often severe enough to affect cognition, thereby adding to the effects of α-synuclein pathology. Tau pathology contributes to dementia in a subset of cases, and amyloid-β pathology confers a worse prognosis, characterised by an accelerated cognitive decline and earlier mortality. A synergistic interaction between α-synuclein, tau and amyloid-β means that they promote one another’s aggregation, leading to a more aggressive disease course. Accordingly, both tau and amyloid-β should be assessed in clinical trials of new disease-modifying therapies targeting α-synuclein, particularly when cognition is included as a study outcome. This will improve the chance of showing efficacy, and reduce the risk of a negative finding driven by the presence of coexistent pathologies.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.
Footnotes
Contributors CS, BC and DG contributed to the planning, conduct and reporting of the work. NM and KG contributed to the conduct and reporting. SG contributed to the reporting.
Funding Funding support for this review was provided by the Neurosciences Foundation (SCO11199; project number 173503).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.