Article Text

Abstract
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive neurodegenerative disease, and only modest disease-modifying strategies have been established to date. Numerous clinical trials have been conducted in the past years, but have been severely hampered by the wide-ranging heterogeneity of both the biological origins and clinical characteristics of the disease. Thus, reliable biomarkers of disease activity are urgently needed to stratify patients into homogenous groups with aligned disease trajectories to allow a more effective design of clinical trial. In this review, the most promising candidate biomarkers in the cerebrospinal fluid (CSF) of patients with ALS will be summarised. Correlations between biomarker levels and clinical outcome parameters are discussed, while highlighting potential pitfalls and intercorrelations of these clinical parameters. Several CSF molecules have shown potential as biomarkers of progression and prognosis, but large, international, multicentric and longitudinal studies are crucial for validation. A more standardised choice of clinical endpoints in these studies, as well as the application of individualised models of clinical progression, would allow the quantification of disease trajectories, thereby allowing a more accurate analysis of the clinical implications of candidate biomarkers. Additionally, a comparative analysis of several biomarkers and ideally the application of a multivariate analysis including comprehensive genotypic, phenotypic and clinical characteristics collectively contributing to biomarker levels in the CSF, could promote their verification. Thus, reliable prognostic markers and markers of disease activity may improve clinical trial design and patient management in the direction of precision medicine.
- ALS
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterised by progressive motor neuron damage. The disease is generally fatal within 2–4 years, but survival can vary largely among individual patients.1 The majority of patients do not present with a family history of ALS and are considered sporadic ALS, while about 10% of cases are familial, and over 50 genes associated with ALS pathogenesis have been identified.
Riluzole remains the only approved therapy in Europe, but treatment prolongs the patient’s life only by a few months. Over 80 clinical trials have been conducted, although their outcomes have been hampered by the wide range of aetiological and clinical features of ALS. Thus, the heterogeneity of the ALS phenotype requires trial designs that stratify patients into more homogenous groups. Previous clinical trials have largely relied on the ALS functional rating scale-revised (ALSFRS-R) or survival as clinical endpoints, which is time-consuming and insensitive to small therapeutic effects.
The importance of biomarkers in therapeutic development has been emphasised in the revised Airlie House consensus guidelines for ALS clinical trials.2 Reliable prognostic biomarkers aid in the stratification of patients and thereby may facilitate early and sensitive detection of therapeutic effects in clinical trials. Moreover, biomarker-based prognostication enables personalised patient management, which is accurately tailored to individual progression rates. Pharmacodynamic biomarkers provide direct feedback on whether a candidate drug achieves the desired effects on its targets and contribute to a deeper understanding of the underlying pathways and mechanisms of action. Additionally, an ALS-specific diagnostic biomarker could be important for early diagnosis and inclusion in clinical trials.
This comprehensive review focuses on prognostic markers and markers of disease activity, with their crucial role in improving therapeutic monitoring. The cerebrospinal fluid (CSF) represents an obvious biofluid source for such biomarkers because of its proximity to the neurodegenerative process.
The PubMed database was searched for the keywords ‘cerebrospinal fluid,’ ‘biomarker’ and ‘amyotrophic lateral sclerosis’ Thus, the most significant and most recent CSF biomarker studies were selected to investigate correlations between biomarker levels and ALS clinical parameters. CSF biomarker studies with purely diagnostic intent were excluded.
Proteomics
Proteomics describes the use of untargeted large-scale analytical approaches to identify proteins that are differentially expressed between groups of samples, and is followed by a validation phase using targeted approaches, such as immunoassays. Recent studies have identified increased levels of glycoprotein non-metastatic melanoma protein B (GPNMB), microtubule-associated protein 2 (MAP2), ubiquitin C-terminal hydrolase-L1 (UCHL1),3 4 capping actin protein (CAPG)3 and cathepsin D4 in the CSF of patients with ALS. GPNMB, UCHL1 and CAPG correlated with the ALSFRS-R score3 while higher CSF GPNMB3 4 and UCHL14 levels predicted shorter survival.
Another longitudinal proteomic approach applied a mixed-effects model of CSF biomarkers that predicted the decline in ALSFRS-R and found significant correlations between inflammatory proteins and disease progression in a small number of patients.5
Limitations in proteomics, such as reduced sensitivity compared with immunoassays, the use of different fractionation and normalisation methods and variations in control groups hamper reproducibility. Nevertheless, some promising biomarker candidates, such as chitotriosidase-1,6 have been discovered using proteomic approaches.
Neurofilaments
Neurofilaments are cytoskeletal proteins that are abundantly expressed in large calibre myelinated axons. Thus, axonal damage causes enhanced leakage of neurofilaments into the CSF, which leads to increased levels of neurofilament light chain (NfL) and phosphorylated neurofilament heavy chain (pNfH). This increase in CSF neurofilaments is more pronounced in ALS than in other neurological disorders,7 8 but their role as diagnostic biomarkers in ALS is limited, as they are not specific to any neuronal type or cerebral region.
In ALS, CSF neurofilament levels appear to most consistently reflect the rate of disability progression related to the speed of neuroaxonal breakdown rather than disease accumulation. Hereinafter, two aspects—rate of progression and accumulated disability—will be discussed in the context of existing studies. An overview of the associations between neurofilaments and ALS disease parameters is provided in table 1.
Association of cerebrospinal fluid neurofilaments with clinical disease parameters reported in patients with ALS
Neurofilaments in the context of accumulated disability
The loss of both upper motor neurons (UMN) and lower motor neurons (LMN) is a hallmark of ALS. The correlation of CSF neurofilament levels with the number of regions showing signs of both UMN and LMN degeneration,9 10 could highlight the role of neurofilaments as markers of neuronal damage. However, this association with combined UMN and LMN loss could not be confirmed for NfL levels in other studies.8 11
Correlation of CSF NfL levels with clinical signs of UMN loss,12–14 and particularly with imaging signs of corticospinal tract degeneration,12 13 further substantiates the notion that degeneration of these large axons, which are abundant in neurofilaments, is accompanied by extensive liberation of NfL. These observations and the increased levels of NfL across many other degenerative and traumatic CNS disorders15 suggest that the rise in CSF NfL represents a downstream effect of neuroaxonal degeneration, rather than an ALS-specific mechanism. Previous studies failed to quantify the relationship between direct measures of LMN damage, such as reduced compound motor action potential amplitudes or cross-sectional nerve area, and CSF NfL levels13 while clinical signs of LMN damage were associated with NfL levels in some studies,11 16 but not in others.17 For pNfH, most studies did not identify an association with the extent of UMN18 or LMN damage,19 but it reportedly correlated with the central motor conduction time as a measure of UMN damage.19
Numerous previous studies have demonstrated correlations between higher neurofilament levels and lower ALSFRS-R scores, which implies a relationship between this biomarker and the absolute degree of disability.7 13 14 16 20–23 However, these analyses were not corrected for the influence of the individual progression speed on neurofilament levels, thus neglecting a common sampling bias caused by the sampling shift. This is known for ALS cohorts, as patients with faster disease progression inevitably experience more extensive functional impairment at the time of sampling than patients with slower progression.8
The findings of higher CSF neurofilaments in UMN dominant patients with ALS compared with LMN dominant phenotypes,13 19 24 corroborate the hypothesis that the degeneration of UMN contributes more strongly to the neurofilament concentration changes. Conversely, UMN-dominant ALS subtypes have longer survival, which, in turn, is associated with lower neurofilament levels. This controversy highlights the complex interplay of factors influencing CSF neurofilament concentrations. Given the multitude of interacting determinants at play contributing to both clinical disease progression and CSF biomarker levels, neither parameter should be considered separately to quantify prognosis. Instead, considering both CSF neurofilament concentration and clinical phenotype in a combined scoring system may enhance precision in stratification of ALS subtypes into more homogenous subpopulations to analyse the effects of new therapeutics in smaller, more homogenous patient groups.
Neurofilaments in the context of rate of disease progression
Generally, a more aggressive disease implies enhanced motor neuronal breakdown per time, with subsequent greater leakage of neurofilaments into the CSF. Thus, CSF neurofilaments may reflect disease activity at early time points, when clinical function is maintained and disability scores remain rather stable. The repeatedly reported correlation between neurofilament levels and disease progression rate underlines this notion.7 9–12 14 16 18 20 21 23 25–27 The speed of clinical disease progression in these studies was calculated based on linear approximations of ALSFRS-R decline, which is critical in terms of the known curvilinear decline of the ALSFRS-R over time.28 Alternative approaches, such as an adapted model of non-linear ALSFRS-R decline (the D50 model), revealed a significant correlation between disease aggressiveness and CSF neurofilament concentrations.8 9 A longitudinal repeated measures analysis of covariance of consecutively obtained ALSFRS-R and the Milano-Torino staging system (MiToS) scores, showed that higher baseline NfL is associated with faster decline of both functional scores.24
Furthermore, higher levels of neurofilaments in the CSF are associated with a shorter time of symptom spreading from spinal or bulbar regions to both (generalisation).16 22 23 Since the combined presence of bulbar and spinal symptoms typically indicates a worse prognosis in ALS cohorts, this points towards the ability of neurofilaments to predict the course of the disease.
Reported associations of lower CSF neurofilament levels with longer survival further support the prognostic role of neurofilaments in ALS.8 11 13 14 16 18 24–27 29–32
Longitudinal assessment of neurofilaments in ALS
The application of neurofilaments as monitoring biomarkers would require stable levels or a predictable temporal profile throughout the disease course. However, owing to the rather invasive nature of collection, studies investigating CSF neurofilaments longitudinally employed rather small cohorts, making conclusions on concentration changes over time challenging. Evidence from longitudinal studies in ALS showed that pNfH remains stable over time,7 9 27 30 proposing pNfH as a candidate biomarker in clinical trials to track therapeutic effects. NfL demonstrated stable levels throughout the disease course in some longitudinal studies,30 33 while others reported slightly decreasing,7 or increasing9 26 concentrations in certain subpopulations of patients with ALS.
Exploration of asymptomatic and symptomatic ALS and FTD gene variant carriers demonstrated that axonal degeneration increases significantly with disease onset and is paralleled by an increase in CSF neurofilament levels.34 35 Increased CSF NfL concentrations up to a year prior to symptom onset have also been reported,33 36 while the duration of the presymptomatic disease stage correlated with the individual speed of disease progression in the symptomatic stage.36 This sheds light on the early biodynamics of the disease and provides biochemical evidence of a presymptomatic disease stage. While it could allow early initiation of clinical trials and even potentially preventive interventions, the known lack of diagnostic specificity represents an obstacle for such approaches.
The inverse associations between CSF neurofilament levels and disease duration found in some studies should be interpreted with caution.7 11 21 22 24 37 While this may indicate declining levels of neurofilaments throughout the course of the disease, it is likely a consequence of the sampling of patients in cross-sectional cohorts. Taking CSF after a longer disease duration is more likely to succeed in patients with a lower speed of disease progression, while CSF samples from patients with highly aggressive disease are usually collected shortly after symptom onset. Instead, for an unbiased, clearer understanding of the temporal profile of CSF neurofilaments, longitudinal CSF studies are essential.
Neurofilaments in the context of ALS-related genetic variants
Lower CSF NfL levels have been reported in patients with ALS linked to SOD1 variants compared with those with wild-type SOD1.31 The most common cause of familial ALS, C9ORF72 hexanucleotide repeat expansion is associated with higher CSF pNfH levels than sporadic ALS.27 30 In the same cohort, CSF pNfH also correlated with progression rate27 30 and survival.27 Thus, the higher CSF neurofilament levels in C9ORF72 ALS are presumably determined by the more aggressive nature of disease in this genetic subtype with faster disease progression, rather than the genetic mutation itself. However, research on this aspect is limited, and further studies on neurofilaments and ALS disease-associated variants should also consider clinical confounders, including disease aggressiveness.
Application of neurofilaments in clinical trials
Due to growing evidence of their potential value as an outcome measure, CSF neurofilaments are increasingly being implemented in clinical trials for ALS. Promising results on exploratory outcome measures have been reported for tofersen, an antisense oligonucleotide treatment for SOD1-mediated ALS. In this phase 1–2 trial, the treatment arm had a milder decline in ALSFRS-R compared with placebo patients, while CSF neurofilament levels decreased in the treated patients but not in the placebo arm.38
Neurofilaments repeatedly proved to reflect the progression speed in ALS, thus likely representing a marker of axonal loss rate over time. Incorporating these biomarkers into clinical trials may aid patient stratification into prognostic subgroups and allow quantitative monitoring of the disease at a biochemical level. Given the evidence of correlation with survival, they may also be considered surrogate markers, thus facilitating the assessment of life-prolonging effects of novel therapeutics.
However, a variety of characteristics have been shown to correlate with CSF neurofilament concentrations, indicating the necessity to consider patient-specific features as a whole. This includes potential intercorrelations of disease measures and sampling confounders when interpreting the CSF concentration of neurofilaments as biomarkers. A possible approach in clinical trials would be patient stratification not only based on neurofilament concentrations, but also based on genotypic, phenotypic and clinical prognostic parameters to ultimately generate more homogenous subgroups for the analysis of treatment effects.
Tau
Tau normally promotes assembly and stability of microtubules in axons, thus increased CSF levels may also mirror neuronal damage. The diagnostic role of this protein in ALS is controversial. While increased total tau (t-tau)16 19 and a reduced phosphorylated (p-)tau/t-tau ratio11 16 39 discriminated ALS from controls in some studies, contradictory findings have been reported.7 37 40 41 Both increased t-tau levels and a reduction of the p-tau/t-tau ratio, correlated with several parameters of disease severity and progressivity, as well as survival, suggesting that these biomarkers may have a prognostic role in ALS (table 2).
Association of cerebrospinal fluid tau proteins with clinical disease parameters reported in patients with ALS
The p-tau/t-tau ratio correlated with MRI signs of corticospinal tract and grey matter atrophy, reflecting the extent of central neuronal damage.13 39 The lower p-tau/t-tau ratio in UMN-dominant ALS phenotypes13 further suggests that this ratio represents central rather than peripheral neuronal damage. The potential of the p-tau/t-tau ratio as a marker of neuronal damage is supported by its reported correlation with CSF NfL.13 Although one study found higher CSF t-tau in patients with bulbar onset than in patients with spinal onset, this may be attributable to the older age of patients with bulbar onset in that cohort40 and was not confirmed in other studies for any of the three tau biomarkers.13 19 37 In a trial of the N-methyl-D-aspartate (NMDA) receptor antagonist memantine, an inhibitor of hyperphosphorylation of tau, CSF t-tau levels declined to the level of healthy controls in some patients, thus potentially reflecting treatment effects.42
Despite some promising clinical correlations, which need further investigation, discrepancies among studies on CSF tau proteins in ALS prevail. Studies including both biomarkers consistently reported superior diagnostic performance of neurofilaments over tau proteins.7 11 16 19 Reasons for the discordant results may be small sample sizes, the variable inclusion of patients with tauopathies as controls, alternative splicing resulting in six different tau isoforms, the multitude of post-translational modifications of the tau proteins and different ELISA kits used. However, the disparity in results may also mirror the known biological heterogeneity of the underlying ALS disease.
Biomarkers of neuroinflammation
There is increasing evidence for the role of neuroinflammation in neurodegenerative disorders, including ALS. However, whether inflammation plays a causative role or occurs as a consequence of neurodegeneration remains unclear. In particular, biomarkers reflecting inflammatory pathways are of special interest with the prospect of immune-targeting therapies for ALS.
Chitinase and chitinase-like proteins
Due to their role in the regulation of immune responses, chitinases have been proposed as biomarkers in numerous diseases and as indicators of microglial and astroglial activation. An increase in chitotriosidase 1 (CHIT1) in the CSF of patients with ALS compared with both disease controls and healthy controls has been reported by unbiased proteomic approaches3 6 32 and by targeted immunoassays.6 11 18 21 29 43–45
Levels of the chitinase-like proteins CHI3L1 (or YKL-40)11 18 32 43 45 46 and CHI3L23 6 18 32 45 are also increased in the CSF of patients with ALS. Different combinations of the three chitinases,32 43 or of CHIT1 with chitinase enzyme activity44 improved the diagnostic performance for ALS in some studies, but the diagnostic accuracy of chitinases alone remained inferior to that of neurofilaments.11 29 Several correlations of chitinases with clinical parameters in ALS have been reported, as summarised in table 3.
Association of cerebrospinal fluid chitinases with clinical disease parameters reported in patients with ALS
CHIT1 and CHI3L1 expression by activated immune cells may directly contribute to motor neuron degeneration, as their concentration in the CSF of patients with ALS correlates with disease progression rate,18 20 21 25 29 32 43 46 severity11 18 21 35 43 44 46 and survival.18 25 29 32 46
Correlation of chitinases with the neuroaxonal damage markers NfL20 21 29 35 46 and pNfH18 21 29 32 35 43 46 provides strong evidence for their direct involvement in neuronal breakdown and their potential to reflect disease activity.
Using a machine learning approach, CSF CHI3L1, α-1-antichymotrypsin and complement factor 1 predicted 49% of the variation in the ALSFRS-R score.47 As all three are synthetised and secreted by microglia and astrocytes, this underscores the crucial role these immune cells play in ALS.
The temporal profile of neuroinflammation in ALS remains a topic of particular interest to better understand its role in ALS. In a cross-sectional analysis, CHIT1 and CHI3L1 levels were normal in asymptomatic carriers of ALS-associated genetic variants, while a sudden increase occurred with symptom onset.35 In this cohort, one SOD1 variant carrier in transition to the symptomatic stage showed normal CSF CHIT1 levels and slightly increased CHI3L1, despite significantly increased NfL levels. This supports the concept that neuroinflammation is a consequence of axonal damage in ALS, rather than a cause. Conversely, in a larger group of individuals with both C9ORF72 and SOD1-mediated forms of ALS an early, CHIT1-associated neuroinflammatory response was observed even in pre-symptomatic patients.45 CSF chitinase levels did not correlate with disease duration and their levels remained longitudinally stable, suggesting a rather constant microglial and astroglial activation in the symptomatic phases of ALS.18 32 43
A duplication in the CHIT1 gene is a common genetic variant in Europe. This polymorphism has been associated with a significantly reduced concentration of CSF CHIT1,35 but was considered a potential confounding factor in biomarker studies, rather than a disease-causing variant for ALS.35
Overall, CHIT1, CHI3L1 and, to a lesser extent, CHI3L2, reflect the progression rate and therefore may serve as prognostic biomarkers in ALS. As markers of microglial and astrocyte activation, they may facilitate the development of anti-inflammatory therapies by monitoring these pathways in clinical trials.
Other chemokines and cytokines
Altered CSF levels of several other cytokines and their correlation with clinical parameters in patients with ALS have been demonstrated in multiple studies in the past years.
Monocyte chemotactic protein 1 (MCP-1), also called CC-chemokine ligand 2 (CCL2) plays an important role in neuroinflammation. Numerous studies have observed an elevation in CSF MCP-1 levels in patients with ALS.30 46 48–52 MCP-1 may reflect disease severity, as its concentration in the CSF correlated with the total Norris scale50 and the ALSFRS-R.48 49 51 MCP-1 concentrations in the CSF are higher in patients with ALS with faster disease progression30 49 51 and shorter survival46 and in patients with C9ORF72-mediated ALS.30 MCP-1 levels were not associated with disease duration,46 50 and are longitudinally stable in consecutive CSF samples.30 Thus, it may be assumed that MCP-1-related inflammatory changes are uniformly present throughout the disease. CSF MCP-1 in patients with ALSs correlated with neurofilament levels and with other pro-inflammatory markers, such as chitinases,46 interleukins (eg, IL-8) or interferon-γ,48 49 underscoring its involvement in neurodegenerative and neuroinflammatory processes in ALS. However, in an independent study, no differences could be seen, so pre-analytical steps might have caused the positive results in other studies.53
Macrophage inflammatory protein-1α and β (MIP-1α and MIP-1β), also called CCL3 and CCL4, respectively, are also members of the CCCL family and may reflect neuroinflammation in ALS, playing a key role in the accumulation of microglia. MIP-1α and MIP-1β levels are elevated in the CSF of patients with ALS.30 48 51 52 54 Both inversely correlate with progression rate48 51 54 and higher levels predicted longer survival.54 55 MIP-1β alone also showed a positive correlation with disease severity, as measured by the ALSFRS-R.48
Soluble CD14 is elevated in the CSF of patients with ALS, especially in those with faster disease progression.56 This presumably originates from membrane-bound CD14 on activated microglia.
Using an unbiased multivariable model to find a panel of CSF and plasma biomarkers that predict survival in patients with ALS, CSF levels of MIP-1β, granulocyte colony stimulating factor (G-CSF), IL-9 and MCP-1 were identified as predictors of longer survival, while IL-5, IL-12 and IL-8 predicted a shorter survival.55
Increased levels of interferon-γ have been reported in patients with ALS. This cytokine was further correlated with the progression and predicted a shorter survival.51 Accordingly, concentrations of interferon-γ are higher in the CSF of more aggressive C9ORF72-mediated ALS relative to SOD1-mediated ALS and other types.57
Multiplex analysis showed significantly increased levels of several ILs and G-CSF in the CSF of patients with ALS, while IL-8 (expressed by activated microglia) was the only cytokine that showed a significant positive correlation with the ALSFRS-R.52 G-CSF was used as a therapeutic agent in a phase 1 trial, where significant reductions in CSF MCP-1 and IL-17 levels following treatment were demonstrated.58
A linear mixed effects model of ALSFRS-R decline to assess the progression rate demonstrated that IL-15 and IL-18 were elevated in fast progressing and C9ORF72-mediated patients with ALS.30 Conversely, IL-10 was associated with better functional status, while IL-4 and eotaxin/CCL11 also demonstrated positive correlations with the disease progression rate.59
Increased CSF concentrations of tumour necrosis factor α (TNFα) have been reported in C9ORF72-mediated ALS relative to other types of ALS30 57 and predicted shorter survival.57 The TNF-related apoptosis-inducing ligand in the CSF of patients with SOD1-mediated ALS was inversely correlated with survival.57 Importantly, this study suggests genotype-specific immune processes in ALS. This underlines the complexity of the disease and may explain the difficulties with reproducibility and validation of biomarkers in different ALS cohorts with incomplete genetic characterisation. Accordingly, a recent study identified elevated CSF IL-6 and soluble IL-6 receptor concentrations in patients with ALS carrying a common genetic variant coding for the IL-6 receptor (IL-6R) compared with patients with ALS without this variant. Importantly, IL-6R variant carriers also showed faster disease progression than other genotypes.60 These findings highlight the multitude of factors influencing CSF cytokine levels, and the need for careful patient characterisation and stratification in clinical trials, as IL-6R blocking therapeutics may play a pivotal role in this genetic subset of patients.60
Further studies are needed to elucidate the underlying mechanisms, but CSF concentrations of the mentioned inflammatory mediators, most of all chitinases, represent promising measures to monitor these pathways and prove to be of prognostic value. Despite rising efforts to investigate neuroinflammatory pathways, it remains unclear whether neuroinflammation represents a primary pathology or a consequence of neurodegeneration. All these markers are non-specific to ALS and are subject to a myriad of cellular interactions. As no single inflammatory mediator accurately represents the complex disease pathology of ALS, a panel of several inflammatory and non-inflammatory markers may be more helpful. Going forward, standardised validation studies of inflammatory CSF biomarkers are desirable to ultimately implement them in clinical trials for neuroinflammation targeting candidate therapeutics.
Biomarkers with neuroprotective role
β-amyloid levels in the CSF of patients with ALS reportedly predict shorter survival61 and correlate with the ALSFRS-R at baseline,41 while the soluble amyloid precursor protein, sAPPβ, was reduced in the CSF of patients with ALS and FTD.25 sAPPβ was further linked to cognitive performance in FTD and the sAPPβ/YKL- 40 ratio was associated with cortical thickness in frontotemporal regions in both the ALS and FTD groups, suggesting that sAPPβ is a biomarker that directly reflects the extent of frontotemporal degeneration.25 Decreases in CSF concentrations of sAPPα and sAPPβ, both known to have neuroprotective properties, have been associated with more rapidly progressive ALS, while the ratio of sAPPα and sAPPβ with pNfH was superior to CSF pNfH alone, in discriminating rapidly progressive patients with ALS from slow progressors and controls.62 Their inverse correlation with CSF pNfH supports the concept that a lower level of these neuroprotective mechanisms fosters faster neurodegeneration in ALS.
Vascular endothelial growth factor (VEGF) is a hypoxia-dependent neurotrophic cytokine that was found to be significantly elevated in the CSF of patients with ALS.48 51 52 VEGF is higher in patients with longer disease duration until first hospitalisation and in patients with limb onset,63 as both are associated with slower disease progression, this may point towards a positive prognostic value of VEGF. Furthermore, VEGF levels are reportedly lower in patients with faster disease progression and shorter survival.51 A lack of VEGF upregulation in hypoxaemic patients with ALS compared with hypoxaemic neurological controls has been observed.64 Additionally, the concentration of VEGF in the CSF positively correlated with paO2 levels in patients with ALS, while the opposite was true for neurological controls, indicating a dysfunction of the response to hypoxia in patients with ALS.64
CSF levels of basic fibroblast growth factor (bFGF) show a similar pattern to VEGF, being increased in patients with ALS compared with controls.51 52 65 bFGF also positively correlated with disease duration65 and survival51 65 and inversely correlated with the disease progression rate.51 65
Ephrin-A5 is a ligand that is predominantly expressed by neurons and binds to the axonal guidance receptor EphA4. In patients with ALS, lower CSF ephrin-A5 concentrations have been associated with shorter survival.66 Further research may broaden our understanding of the neuroprotective properties of ephrins and their involvement in ALS. Meanwhile, the EphA4 receptor already represents a promising therapeutic target,67 and clinical trials of such treatments could benefit from an adequate biomarker to monitor target engagement.
A phase 2 clinical trial of mesenchymal stem cell-neurotrophic factor cells used CSF levels of inflammatory and neurotrophic factors to monitor target engagement.68 Increased expression of the neuroprotective markers VEGF, hepatocyte growth factor and leukaemia inhibitory factor and a decrease in neuroinflammatory markers MCP-1 and stromal cell-derived factor-1α were demonstrated post-treatment.68
Overall, these observations support the hypothesis that in ALS, an interplay of neuroprotective and neuroinflammatory factors modulates disease progression and accentuates the potential of both neuroinflammatory and neuroprotective CSF biomarkers to directly monitor treatment effects in clinical trials.
Cytoplasmic protein hallmarks
TDP-43
Neuronal and glial cytoplasmic inclusions of phosphorylated transactive response DNA-binding protein of 43 kDa (TDP-43) represent a neuropathological hallmark of ALS. Several studies have reported elevated CSF TDP43 levels in patients with ALS.69 There is some controversy about the specificity of these assays for the brain-derived pathological form of TDP-43, as several modified forms of this protein exist.70 Nevertheless, given its important role in disease pathology, TDP-43 represents a promising specific biomarker for ALS and FTD and a deeper understanding of its modified forms, their origins and interactions are crucial for future research.
Dipeptide repeats
Poly(GP) is a dipeptide repeat protein translated from the expanded intronic hexanucleotide repeat sequence in C9ORF72, the most common hereditary cause of ALS. It was elevated in the CSF of individuals carrying the expansion71 and increased CSF poly(GP) in asymptomatic carriers may indicate that poly(GP) is secreted from viable neurons, rather than passively released from degenerating neurons.71 72
Both TDP-43 and poly(GP) are of particular interest because of their specificity and direct relation to ALS pathology. As promising therapeutic targets,72 73 biochemical monitoring of these proteins in the CSF may aid in the development of drugs targeting these inclusions. However, challenges with immunospecificity and the intracellular origin of TDP-43 need to be considered. However, to date, evidence of their association with disease activity or progression is lacking. This is an intriguing question for future ALS biomarker studies, given their neuronal origin and specificity for the disease.
Markers of oxidative stress
The licencing of the free radical scavenger drug edaravone for ALS in the USA and Japan sheds light on the role of oxidative stress as a target for novel therapies. A reduction in the oxidative stress marker 3-nitrotyrosine to nearly undetectable CSF levels was observed following treatment with edaravone.74 Similarly, the CSF marker of anti-oxidative activity OXY was significantly reduced in patients with ALS compared with controls, which improved on treatment with edaravone and also correlated significantly with clinical functional scores.75 Neither biomarker was included in the Pivotal efficacy trial.76
In a phase 1–2 trial of the antisense oligonucleotide treatment (tofersen) for SOD1 ALS, treated patients showed significant decreases in CSF SOD1 levels.38 Another measure of oxidative stress, the oxidation reduction potential (ORP), exhibited a significant negative correlation with the ALSFRS-R score, indicating an increase in ORP with worsening of functional impairment.77
Arginine methylation is an important method for monitoring RNA processing, including transcription and translation. The ratio of asymmetric dimethyl L-arginine (ADMA) and L-arginine plays an important role in oxidative stress, as arginine serves as a source of nitric oxide (NO), while ADMA inhibits NO synthase. The ADMA/L-arginine ratio did not demonstrate significant differences between the ALS and control groups, but it correlated with the progression rate and respiratory status and predicted poor survival in patients with ALS with a higher sensitivity than the respiratory function.78 While the role of this ratio in the pathogenesis of ALS requires further investigation, these findings propose the ADMA/L-arginine ratio as a biochemical measure of disease progression and a predictor of survival.
Outcome parameters in biomarker studies
Tremendous efforts in biomarker research for ALS in the past years have resulted in numerous promising CSF biomarker candidates, showing correlations with ALS disease parameters (figure 1).

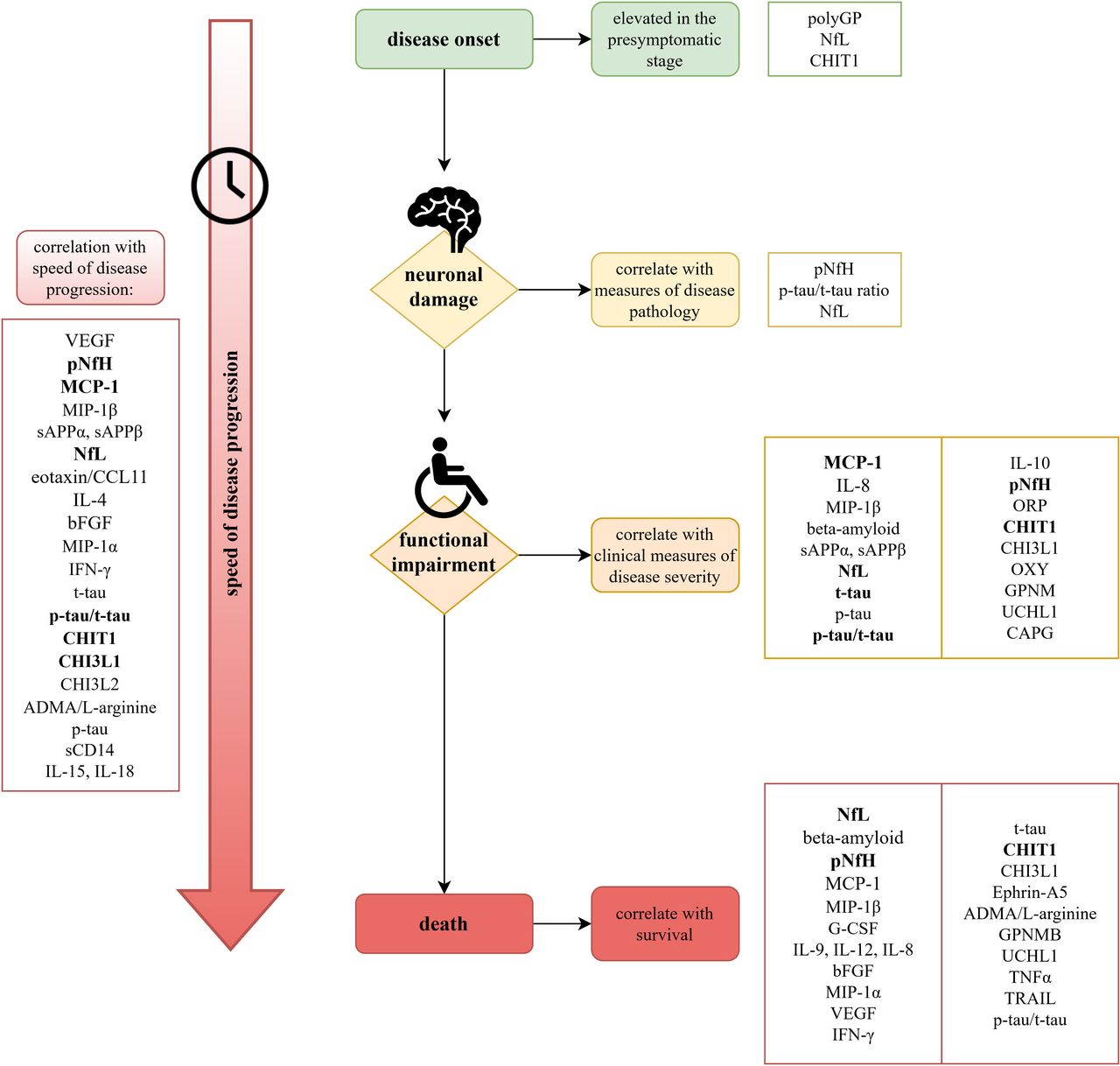{kind=link}
Correlation of CSF biomarkers with individual disease parameters in patients with ALS. The figure displays a timeline of different outcome measure categories and associated biomarkers. Correlations, which have been reported in three or more studies discussed in this review are given in bold. However, the authors would like to emphasise that the number of studies reporting an association is not the sole indicator of importance, as some rather newly discovered biomarkers inevitably need more time to gather broad evidence. The biomarkers are therefore also given in the order of first publication of a related study, with those reported first at the top. Detailed discussion of each biomarker’s potential can be found in the text. ALS, amyotrophic lateral sclerosis; ADMA, asymmetric dimethyl L-arginine; bFGF, basic fibroblast growth factor; CCL3, chemokine ligand 3; CHI3L, chitinase-like proteins; CHIT1, chitotriosidase 1; G-CSF, granulocyte colony stimulating factor; GPMNB, glycoprotein non-metastatic melanoma protein B; IFN, interferon; IL, interleukin; MCP-1, monocyte chemotactic protein 1; MIP-1α, macrophage inflammatory protein-1α; NfL, neurofilament light chain; pNfH, phosphorylated neurofilament heavy chain; p-tau, phosphorylated tau; sCD, soluble CD; sAPP, soluble amyloid precursor protein; t-tau, total tau; TNF, tumour necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; UCHIL1, ubiquitin C-terminal hydrolase-L1; VEGF, vascular endothelial growth factor.
Previous clinical trials have largely relied on indirect and time-consuming endpoints, such as survival and ALSFRS-R decline over time. Therefore, biomarkers that reflect progression and severity may be of immense value by shortening trial duration and enabling stratification of patients into more homogenous subgroups. However, many biomarker studies conducted so far have disregarded the impact of intercorrelations between clinical parameters that occur in most ALS cohorts. Due to the sampling shift, patients with faster disease progression inevitably experience more extensive neuronal damage at the time of investigation. Therefore, the functional status at the time of sampling is highly dependent on the speed of disease progression. This underlines the necessity of multivariate analyses when investigating correlations between clinical parameters and CSF analytes. These should incorporate possible confounders, such as progression speed and disease severity, as well as age and gender or pre-analytical factors.
Several CSF biomarkers have been demonstrated to be associated with the ALSFRS-R as an indicator of disease severity. However, the application of the ALSFRS-R has several limitations, including its multidimensionality, non-linearity and floor effects. Clinical milestones could instead display more appropriate measures of disease severity, as implemented in the King’s staging or MiToS staging systems.79 These staging systems still need to be more regularly applied in CSF biomarker studies to explore their potential.
As a measure of progression speed, the majority of studies have used the disease progression rate calculated as (48–ALSFRS-R)÷disease duration in months. This presumes a linear decline of the ALSFRS-R, despite the observation of its curvilinear decline, which has the potential to distort results.28 A large variety of CSF biomarkers, first and foremost neurofilaments, but also chitinases, CHI3L and other inflammatory markers correlated with this linear progression rate and therefore presumably reflect disease activity. Two studies applied an individualised sigmoidal function of ALSFRS-R decline, the D50 disease progression model. They demonstrated correlations of pNfH and NfL with the modelled parameter D50, representing the time for a patient to reach halved functionality and thus describing patients’ overall disease aggressiveness.8 9 This model, is based on the ALSFRS-R, but also takes into account its sigmoidal decline, and is a promising new approach considering the widespread use of the ALSFRS-R, which facilitates further retrospective and validation studies of the model. The application of such models, providing a reliable quantifiable framework of clinical disease progression, may propel the disclosure of clinical correlations of candidate biomarkers.
Survival, as an endpoint in clinical trials, is an essential indicator of treatment success but requires long study durations, while potentially being biased by many confounding factors. Therefore, a biomarker reflecting this endpoint may only be partly related to disease activity itself. Nevertheless, it may aid in planning therapeutic management and guide the development of life-prolonging therapeutic candidates. A large number of CSF biomarker studies have demonstrated correlations with survival, and the largest evidence exists for neurofilaments and chitinases.
Finally, a pharmacodynamic biomarker should reflect disease activity while remaining longitudinally stable. In clinical trials, changes in the CSF concentration of such markers can serve as early and specific indicators of drug efficacy. Thus, pharmacodynamic biomarkers may save precious time, resources and money in phase 2 trials by refuting drugs without expected effects. Conversely, propelling promising candidates for phase 3 trials and guiding the search for an appropriate dose-effect relationship. In fact, considering the fiasco of clinical trials in the past, it is recommended that every new drug should prove its effect on the target through a pharmacodynamic biomarker. This is essential not only for the examined drugs, but also for a better understanding of drug effects in general. However, the identification of such markers requires large longitudinal CSF studies, which are disappointingly scarce for ALS.
Conclusions and future directions
Disease activity biomarkers are urgently needed to propel the development of disease-modifying therapies for ALS. Therefore, a reproducible correlation of biomarker concentrations with either clinical status or disease progression speed is of paramount importance.
Despite immense efforts in biomarker research and the discovery of several candidate molecules that have been repeatedly shown to reflect disease aggressiveness or prognosis, none of these markers has reached routine applicability in clinical practice. Most CSF biomarker studies are rather small and single centred. They use non-standardised methods, and clinical outcome parameters vary between studies, hampering comparability.
As our knowledge about disease mechanisms and genetics broadens, biomarker analyses need to employ well-defined ALS cohorts, bearing in mind that CSF biomarker profiles are influenced by a multitude of factors and may differ among certain subgroups of patients with ALS. As no single disease-causing pathological mechanism for ALS has yet been identified, but rather a number of synergistic, interacting mechanisms, a combination of biomarkers displaying different pathways may be most accurate in reflecting disease activity and prognosis. This could lead to future multidrug trials and individualised precision medicine for effectiveness subgroups.
Additionally, international, large databases for ALS may expedite the research process and augment the insights gained from a limited number of patients with ALS, allowing retrospective analysis and comparative analysis of several promising biomarker candidates.
Going forward, multivariate analyses and reliable quantification of disease trajectories, such as recently proposed models for ALSFRS-R decline, could enhance precision in biomarker studies and aid in confirming correlations of promising biomarker candidates with individual disease metrics. A standardised choice of clinical endpoints may also enhance the reproducibility of clinical correlations with candidate biomarkers, ultimately accelerating the incorporation of established prognostic and monitoring CSF biomarkers into clinical trials.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
References
Footnotes
MD and RS contributed equally.
Contributors MD, RS and JG contributed to the initial conception of the study. MD wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding The present study was supported by the motor neuron disease association (MNDA), Deutsche Gesellschaft fuer Muskelkranke (DGM), BMBF-JPND networks Genfi-Prox (01ED2008A), MBF-Era-net moodmarker (01EW2008), the German Federal Ministry of Education and Research (FTLDc 01GI1007A), Thierry Latran Foundation (D.6438) and ALS association (D.5809). MD was supported by the ERASMUS programme of the European Union. RS is supported by the Deutsche Forschungsgemeinschaft (DFG) with a clinician scientist programme (WI 830/12-1).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.