Article Text

Abstract
Background: While spasticity is commonly treated with oral agents or botulinum neurotoxin (BoNT) injection, these treatments have not been systematically compared.
Methods: This study performed a randomised, double-blind, placebo-controlled trial to compare injection of BoNT-Type A into spastic upper limb muscles versus oral tizanidine (TZD), or placebo, in 60 subjects with upper-limb spasticity due to stroke or traumatic brain injury (TBI). Wrist flexors were systematically injected, while other upper limb muscles were injected as per investigator judgement. Participants were randomised into three groups: (1) intramuscular BoNT plus oral placebo; (2) oral TZD plus intramuscular placebo; (3) intramuscular placebo plus oral placebo. The primary outcome was the difference in change in wrist flexor modified Ashworth score (MAS) between groups. Other outcome measures included MAS at elbow and finger joints, Disability Assessment Scale (DAS) and adverse events (AE).
Results: BoNT produced greater tone reduction than TZD or placebo in finger and wrist flexors at week 3 (p<0.001 vs TZD; p<0.02 vs placebo) and 6 (p = 0.001 vs TZD; p = 0.08 vs placebo), and greater improvement in the cosmesis domain of the DAS at week 6 (p<0.01). TZD was not superior to placebo in tone reduction at either time point (p⩾0.09). The incidence of AE related to study treatment was higher with TZD than in the BoNT (p<0.01) or placebo groups (p = 0.001).
Conclusions: BoNT is safer and more effective than TZD in reducing tone and disfigurement in upper-extremity spasticity, and may be considered as first-line therapy for this disorder.
Statistics from Altmetric.com
Stroke occurs in approximately 700 000 people in the USA each year,1 leaving many survivors with motor impairments, including spasticity. Spasticity involving the upper limb may be particularly disabling.2 3 Before the introduction of botulinum neurotoxin (BoNT) as a chemotherapeutic agent, the treatment of spasticity relied predominantly upon systemic CNS neurotransmitter agonists or antagonists such as baclofen or tizanidine (TZD).4 5 However, their utility is limited by sedation and other side effects associated with their systemic distribution and non-selective mechanisms of action. More recently, focal intramuscular injection of BoNT has been shown to be safe and effective in the treatment of spasticity.6–12 Still, most physicians continue to initiate therapy with oral agents due to their lesser short-term cost or perceived convenience, or in response to payor mandate.13 However, there has been no controlled study that directly compares focal and systemic agents in patients with spasticity.
METHODS
This study was a randomised, controlled, parallel group, double blind, multicentre study of the efficacy, safety and tolerability of BoNT-A (Botox, Allergan, Irvine, California) injection into upper-limb muscles versus oral TZD (Zanaflex, Acorda Therapeutics, Hawthorne, New York) in subjects with upper-limb spasticity due to stroke or TBI.
Eligibility and enrolment
Study population
Eligible participants were 18–85 years of age, with prior stroke (cerebrovascular accident with a neurological deficit persisting at least 24 h) or traumatic brain injury (TBI) ⩾ 3 months earlier, and spasticity of the wrist, as demonstrated by a score of ⩾3 for wrist flexor tone on the modified Ashworth Scale (MAS),14 with 0 indicating normal tone and 5 rigid flexion. An additional criterion for enrolment was difficulty with hygiene or dressing, pain or malposition of the wrist, as evidenced by a score of ⩾2 on the Disability Assessment Scale (DAS).2 One domain was chosen by the investigator and the participant or care giver as the Principal Therapeutic Target (PTT) as assessed at the time of initial screening. A score of 0 on the DAS indicates no disability, and 3 is severe disability.
Exclusion criteria included severe contracture at the wrist (inability to passively move the joint by >10°); prior tendon transfer; prior phenol/alcohol nerve block in the study limb; BoNT injection into the target limb within 4 months; prior casting of the study limb within 2 weeks; severe muscle atrophy or infection in target sites; orthostatic hypotension or treatment with oral antispasticity agents within 14 days; impaired renal or hepatic function; or current anticoagulant therapy with INR⩾3.5. Women were excluded if they were pregnant or planning to become pregnant during the course of the study. Participants taking other CNS medications, (eg, antidepressants), were required to be on a stable dose for ⩾2 months previously. Physical/occupational therapy, if used, was required to be maintained unchanged throughout the study.
The study was consistent with Good Clinical Practice guidelines and applicable regulatory requirements, and was approved by the IRB at all sites. Written informed consent was obtained from all participating subjects.
Randomisation scheme and treatment
Patients were randomly assigned to receive treatment with one of three treatments: group 1, intramuscular BoNT-A plus oral placebo; group 2, intramuscular placebo plus oral TZD; group 3, intramuscular placebo plus oral placebo. TZD was supplied as 4 mg tablets. The dose of the blinded oral study medication (TZD or placebo) was initiated at 2 mg/day to a maximum of 36 mg/day. The oral study medication was taken twice per day, and titrated by 4 mg increments every 3–4 days as per telephone contact between the subject and study nurse/investigator. If a subject tolerated all dose increases, a maximum dose of 36 mg could be reached by day 27–28. Any subject experiencing side effects was instructed to return to the previous tolerated dose and maintain it for three more days. Slower titration then occurred at 2 mg increments every 3–4 days. If the subject again experienced any side effects, they returned to the previous tolerated dose and maintained it until the end of the treatment period (visit 6, week 18).
Each subject received an injection of BoNT-A or saline placebo at visit 2. Each phial of Botox contains 100 units (U) of BoNT-A, 0.5 mg of human albumin and 0.9 mg of sodium chloride in a sterile, vacuum-dried form without preservative.
In order to maintain blinding, an individual other than the injecting and evaluating investigators prepared the phials for injection. BoNT-A was diluted with preservative-free normal saline. Based on our experience with BoNT-A volume/potency studies, suggesting a greater effectiveness of high-volume/dose injections in larger muscles,15 lyophilised Botox, 100 units (U)/phial, was reconstituted with 5 cm3 of preservative-free saline for injections in muscles above the elbow (20 U/cm3) and with 2 cm3 of saline for muscles below the elbow (50 U/cm3). All subjects were required to receive a standardised dosage of Botox of 50 U (1.0 cm3)/muscle into each of the wrist flexors (flexor carpi radialis and ulnaris). The remainder of the affected upper-extremity muscles, from the shoulder to fingers, could be injected as per the investigator’s discretion, based on the subject’s disability, to a maximum total dose of 500 U. Injections employed a needle stimulation technique, with a monopolar injection electrode.16 Once the target muscle was identified, by obtaining an appropriate contraction with the lowest possible stimulus intensity, BoNT was injected into one to four sites, based on the size of the muscle.
The study duration was 22–24 weeks and consisted of a 1-day to 2-week screening period (visit 1), an injection and oral treatment initiation visit (visit 2/baseline visit) and follow-up visits at weeks 3, 6, 12 and 18. At the end of the treatment period, subjects were monitored for a further 4 weeks.
Assessments
The primary efficacy measure was the difference in change from baseline in the wrist MAS between treatment groups at visit 4 (6 weeks post-treatment initiation). Secondary outcome variables included differences in change from baseline in the following measures: DAS, Modified Frenchay Scale,17 10 m walking speed, contralateral grip strength, finger tap test, Epworth Sleepiness Scale and cognitive evaluations (Geriatric Depression Scale, Questionnaire and Letter–Number Sequence). Other variables included a comparison of the number of subjects that discontinued oral study medication and those that required a slower titration schedule of oral study medication between treatment groups. Safety assessments included adverse events, vital signs, physical examination and liver function tests.
Statistical analysis
To detect a mean difference between BoNT and TZD of 0.75 on the primary efficacy variable, MAS change at the wrist from baseline to week 6, considering a two-sided test with alpha of 0.05 and power of 0.80 and a standard deviation of 0.8, a sample size of 20 subjects per treatment group was required (total 60 patients). A 0.75 change on the MAS was considered clinically significant.2 Primary outcome and safety analyses were conducted on an intent-to-treat basis. Missing values were not replaced. Changes from baseline in MAS and DAS were compared at all postbaseline visits between all three treatment groups using rank ANCOVA, with the baseline value as a covariate. If the overall comparison revealed any significant differences across groups, pairwise comparisons were performed.
RESULTS
Patient characteristics
Sixty patients were randomly assigned for treatment at 10 centres (20 BoNT, 21 TZD, 19 placebo; fig 1). Treatment groups were balanced for baseline wrist MAS score, age, gender and race (table 1).
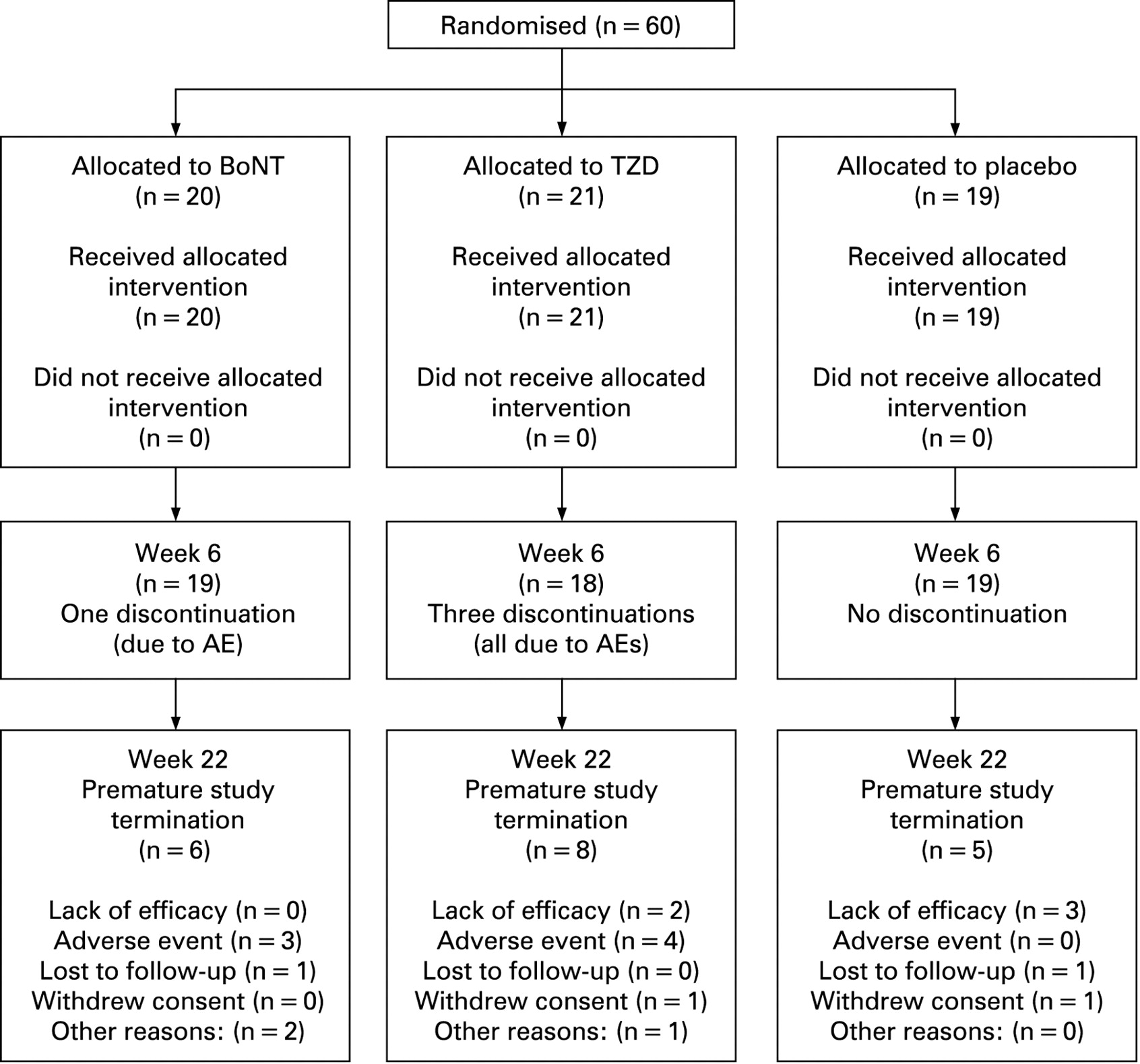
Consort flow diagram of subject disposition. AE, adverse event; BoNT, botulinum neurotoxin; TZD, tizanidine.
Subject disposition
Overall, 56 subjects (93.3%) remained in the study to the time of the primary endpoint at week 6 (19 BoNT (95%); 18 TZD (86%), 19 placebo (100%)); 41 patients (68%) completed the study to week 22; six BoNT subjects (30%), eight TZD (38%) and five placebo subjects (26%) terminated early (fig 1). The dose of BoNT and equivalent injected placebo was similar in the groups (mean (SD) U; BoNT: 392.0 (139.3), TZD: 414.0 (111.5), placebo: 371.6 (134.9); p = 0.64). The titrated dose of TZD was lower in the TZD group (table 1) at week 6 (mean mg (SD); BoNT: 30.0 (12.3), TZD: 20.0 (12.1), placebo: 29.8 (12.5); p = 0.02) and week 18 (BoNT: 22.1 (15.5), TZD: 14.7 (13.5), placebo: 32.6 (9.5); p = 0.007).
Efficacy
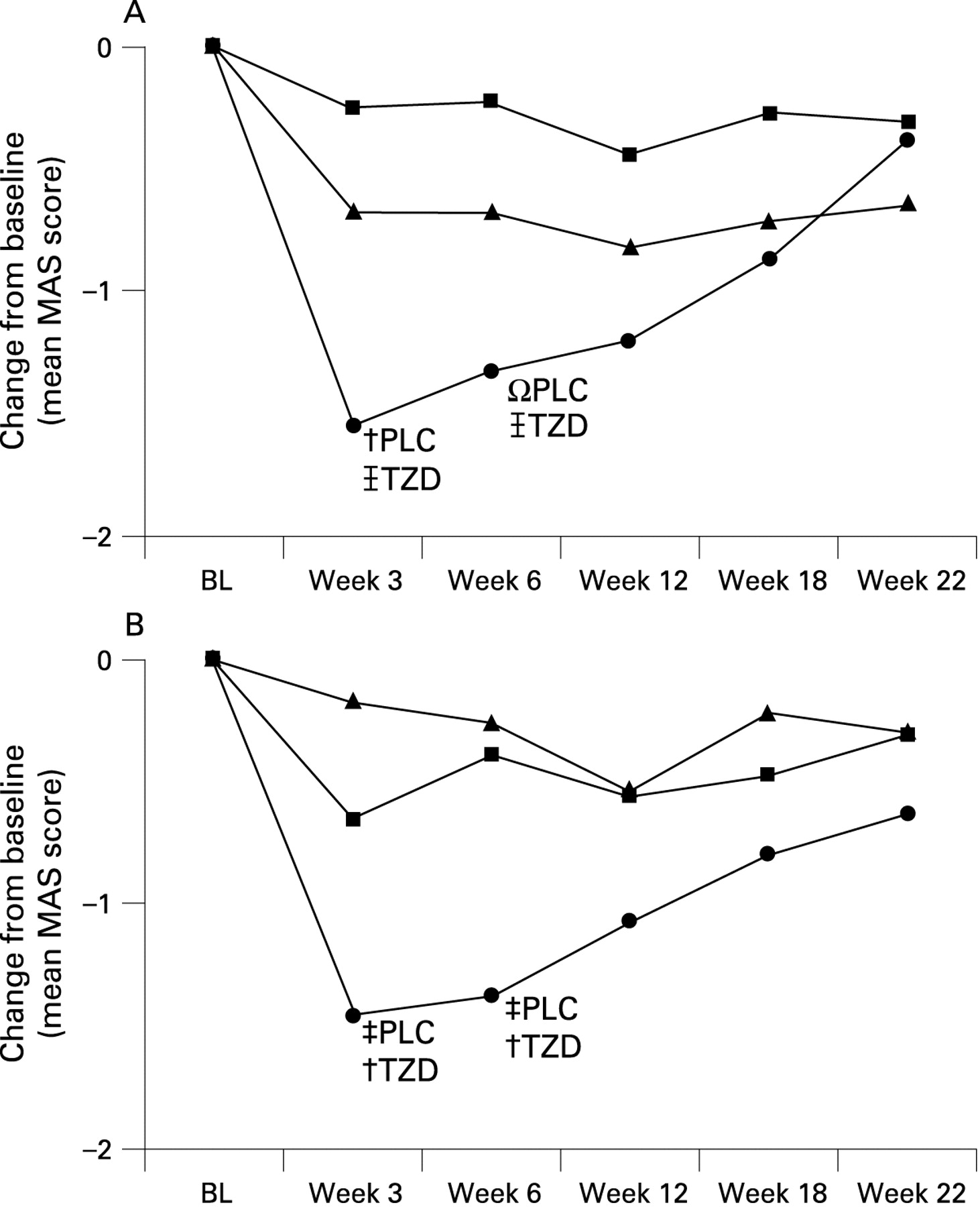
BoNT produced a greater reduction in wrist flexor tone than TZD or placebo at week 3 (mean change from baseline in MAS: BoNT: −1.55 (1.19); TZD: −0.25 (0.64), placebo: −0.67 (0.91); p = 0.001; BoNT vs TZD: p<0.001; BoNT vs placebo: p = 0.02; ZFX vs placebo: p = 0.09; fig 2A) and week 6 (BoNT: −1.32 (0.89); TZD: −0.22 (0.88), placebo: −0.68 (1.00); p<0.01; BoNT vs TZD: p = 0.001; BoNT vs placebo: p = 0.08; ZFX vs placebo: p = 0.12). Wrist tone increased at weeks 12 and 18. There was a higher rate of wrist flexor tone improvement of at least one grade at week 6 in the BoNT group (n = 16 (84.2%)), as compared with TZD (5 (27.8%)) and placebo (12 (63.2%); p = 0.002).

Treatment response in mean flexor tone effect over time. Modified Ashworth Score (MAS): botulinum neurotoxin (•) produced a greater reduction in both wrist (A) and finger flexor (B) tone than tizanidine (TZD; ▪) and placebo (PLC; ▾) at weeks 3 and 6 (Ωp<0.1; †p<0.05; ‡p<0.01;  p<0.001). BL, baseline.
p<0.001). BL, baseline.
BoNT also produced a greater reduction in finger flexor tone than TZD or placebo at weeks 3 (BoNT: −1.45 (1.19); TZD: −0.65 (0.75), placebo: −0.17 (0.71); p = 0.001) and 6 (BoNT: −1.37 (1.46); TZD: −0.39 (0.98), placebo: −0.26 (0.93); p<0.02) (fig 2B). In this study, investigators had the discretion to select the dose used for injection of finger flexors; injection of higher BoNT doses resulted in a greater reduction in finger flexor tone than lower doses (fig 3). In this regard, the observed reduction in finger flexor tone after BoNT injection was greater than in the wrist flexors. The mean MAS reduction at the finger flexors for patients receiving any dose of BoNT into the finger flexors was −1.63 (1.20) at week 3 and 1.53 (1.41) at week 6. The corresponding improvements for those receiving ⩾100 U of BoNT into the finger flexors was 2.00 at week 3 (p = 0.001 vs TZD, p<0.001 vs placebo) and 1.62 at week 6 (p = 0.01 vs TZD, p<0.02 vs placebo); for those with injection ⩾200 U, the mean MAS reduction was 2.75 at both week 3 (p = 0.03 vs TZD, p = 0.01 vs placebo) and week 6 (p = 0.08 vs TZD, p<0.02 vs placebo).

{kind=link}
{kind=link}
{kind=link}
Mean tone effect in patients injected in the finger flexors: all, only ⩾100 U and only ⩾200 U botulinum neurotoxin (BoNT) doses. Modified Ashworth Score (MAS): BoNT injections (•) in the finger flexors produced a greater reduction in finger tone than tizanidine (TZD; ▪) and placebo (PLC; ▾) at weeks 3 and 6 (Ωp<0.1; †p<0.05; ‡p<0.01).
There was also a higher rate (p<0.04) of finger flexor tone improvement of at least 1 grade at week 6 in the BoNT group (n = 14 (73.7%)), as compared with TZD (7 (38.9%)) and placebo (7 (36.8%)). There were no tone-reduction differences between groups in elbow flexors and pronators, and shoulder extensors.
For the total cohort, the choice of the PTT on the DAS was dressing (32.2%), hygiene (28.8%), cosmesis (23.7%) and pain (10.2%), with the remainder being combinations of these categories. There were no group differences at baseline in PTT (mean = 2.42 (0.76)). There was a non-significant trend to a greater reduction in PTT at week 6 in the BoNT group (BoNT: −1.13 (1.08); TZD: −0.47 (1.18); placebo: −0.67 (1.08); p = 0.20). The cosmesis domain of the DAS improved at week 6 in the BoNT group as compared with TZD and placebo (BoNT: −1.00 (1.00), TZD: + 0.12 (0.93); placebo: −0.16 (1.01), p<0.003). There were no group differences in other domains of the DAS, or in other secondary measures. Analysis of the Modified Frenchay Scale is ongoing and will be reported separately.
Safety and tolerability
The incidence of adverse events (AEs) related to study treatment was higher in the TZD group than in the BoNT or placebo groups (p<0.01), and no different between the BoNT and placebo groups (p = 0.43) (table 2). The most common AE was somnolence (TZD: 48%, BoNT: 20%; placebo: 10.5%). Elevated liver enzymes occurred in 10% of the TZD group, with none in the BoNT and placebo groups.
DISCUSSION
Paradigms for the treatment of poststroke spasticity and TBI often include oral medications before BoNT injection.5 13 However, the absence of comparative trials between BoNT and oral medications can make the choice of initial treatment difficult. This study demonstrates that focal injections of BoNT are more effective at decreasing muscle tone in the upper extremity than oral TZD or placebo. BoNT resulted in a greater reduction in tone at the finger and wrist flexors than those treated with TZD.
Unlike the findings in the wrist and finger flexors, there was no change in spasticity in the elbow or shoulder muscles treated. This finding may result in part from BoNT underdosing or too few subjects injected in these muscles. While wrist flexors needed to be injected, other muscles were treated at the discretion of the investigator, and proximal upper-extremity muscles were not treated systematically.
This study demonstrated that TZD was ineffective in decreasing muscle tone in patients with poststroke upper-extremity spasticity. This result may be due to inadequate dosing, limited by the occurrence of adverse effects. The lack of efficacy of TZD at 6 weeks was demonstrated in all muscles evaluated and stands in contrast to studies of TZD in spasticity due to spinal cord injury.18 19 It is not surprising that patients with brain disease may be more sensitive to CNS side effects of TZD than those with spinal cord pathology.20 21
Studies have generally failed to demonstrate improved active function after treatment with any antispasticity medication. Global scales developed as measures of quality of life and stroke recovery are not sufficiently sensitive to detect changes in a single limb after treatment.6 17 Studies of oral medications usually focus on overall function and well-being. The DAS was developed in response to changes noted in patients treated for focal upper-limb spasticity.2 A self-report measure, the DAS measures changes in selected daily activities using the upper extremity rather than a global or overall quality-of-life scale. The inclusion of the DAS in this study permitted assessment of change in key domains of hand function, such as hygiene and cosmesis. The results of this study suggest that patients’ perception of limb position (cosmesis) was improved. There was a trend toward an improvement in the primary therapeutic target at week 6.
While this is the only study to directly compare BoNT and an oral anti-spasticity medication, the three study arms had only 20 subjects each, and investigators were required to treat only the wrist flexors, with the remainder of the muscles at their discretion. This may account for the lack of change in proximal muscles. Our findings are similar to a study of 126 patients with upper-extremity poststroke spasticity treated with BoNT or placebo.2 In that study, there were changes in the DAS in those subjects receiving active treatment in addition to changes in muscle tone.
The current study was not powered to determine changes in the DAS. With only 20 subjects receiving active BoNT, it is doubtful that changes in DAS would be seen, given this study design. However, the trend indicates that the DAS may be a useful tool in larger trials. While the inclusion of a larger number of subjects in a trial such as this would increase the likelihood of detecting active functional improvement, it is difficult to recruit subjects into placebo-controlled trials of therapeutic agents which are currently marketed for the treatment of spasticity. Further analysis of the Modified Frenchay Scale data in our trial will indicate if this is a sensitive measure of functional improvement.
This study demonstrated increased adverse effects with TZD when compared with BoNT. In a trial comparing trihexyphenidyl with BoNT-A in cervical dystonia, changes on the Disability section of the Toronto Western Spasmodic Torticollis Rating Scale Tsui Scale and the General Health Perception Subscale favoured BoNT.22 In these patients, adverse effects were less frequent in the BoNT group than in those receiving trihexyphenidyl. Prior to this study, anticholinergics were considered first-line therapy for cervical dystonia.23 The current study of BoNT compared with TZD for spasticity reveals similar results to those in the CD study, with more adverse events in the oral treatment group.
While the results of this study cannot be extrapolated to other drugs beyond TZD and BoNT-A, the low incidence of side effects in the injection group (both placebo and BoNT) in comparison with TZD suggests that injections alone are well tolerated in this population. In addition, the systemic side effects in the TZD group are concerning in a population that may already be compromised due to neurological disability. As treatment paradigms are developed for pharmacological treatment of spasticity, they should select the most effective drug with the fewest side effects as first-line therapy. The results of this study suggest that injections of BoNT alone to treat focal or multifocal spasticity decrease muscle tone with few systemic effects and should be considered as the primary treatment before oral medications.
Acknowledgments
BoNT/TZD Study Team: Mount Sinai Medical Center, New York (Study Chair: D Simpson, J-M Gracies; M-C George, Alexandra Nmashi); University of Rochester, Rochester, New York (R Barbano, D Berry); Indiana University School of Medicine, Indianapolis, Indiana (A Brashear, L Kuhn); Burke Rehabilitation Hospital, White Plains, New York (P Fonzetti, M Montalvan); Neurology Group of Bergen County, Ridgewood, New Jersey (K Levin, S Pipala, L Warnock); General Neurology and Movement Disorders, New Hyde Park, New York (M Forrest Gordon, E Licari, N Brown, D Fury-Cesarski); Methodist Rehabilitation Center, Jackson, Mississippi (S Yablon, P Russum); North Staffordshire Rehabilitation Centre, University Hospital of North Staffordshire, Stoke on Trent, UK (A B Ward, S Javaid); University of Kentucky Medical Center, Lexington KY (K Nelson, L C Pettigrew, D Taylor). The authors thank M Jensen and N LeGall of Ethica Clinical Research, Inc, and M C George, for their invaluable assistance. The results of this study were presented in part at the 2007 Annual Meeting of the American Academy of Neurology.
REFERENCES
Footnotes
Competing interests: DMS and JMG received consultancy fees and research grants from Allergan, Solstice and Merz; SAY received consultancy fees from Allergan, Merz, and Athena Neurosciences and research grants from Athena, and Medtronic; RB received consulting fees, educational grants and speaking honoraria from Allergan and a research grant from Merz; AB received consultancy fees from Merz and research grants from Allergan, Ipsen and Merz, and provided expert testimony related to botulinum toxin.
Funding: DMS of Mount Sinai School of Medicine is the sponsor of the study. The study was funded by an unrestricted grant by Allergan, Inc. Allergan had no influence on the design, interpretation or reporting of the study.
Clinical trial registration: This study is registered at http://clinicaltrials.gov/:NCT00430196.
Ethics approval: Ethics approval was provided by the following Institutional Review Boards: Mount Sinai Medical Center, New York, NY University of Rochester, Rochester, NY Indiana University School of Medicine, Indianapolis, IN Burke Rehabilitation Hospital, White Plains, NY Neurology Group of Bergen County, Ridgewood, NJ Long Island Jewish Medical Center, New Hyde Park, NY Methodist Rehabilitation Center, Jackson, MS North Staffordshire Rehabilitation Centre, University Hospital of North Staffordshire, Stoke on Trent, UK University of Kentucky Medical Center, Lexington KY.
Patient consent: Obtained.
See Editorial Commentary, p 359
Linked Articles
- Editorial commentary