Article Text

Abstract
Background Although deep brain stimulation (DBS) of the subthalamic nucleus (STN) is a highly effective therapeutic intervention in severe Parkinson's disease, its mechanism of action remains unclear. One possibility is that DBS suppresses local pathologically synchronised oscillatory activity.
Methods To explore this, the authors recorded from DBS electrodes implanted in the STN of 16 patients with Parkinson's disease during simultaneous stimulation (pulse width 60 μs; frequency 130 Hz) of the same target using a specially designed amplifier. The authors analysed data from 25 sides.
Results The authors found that DBS progressively suppressed peaks in local field potential activity at frequencies between 11 and 30 Hz as voltage was increased beyond a stimulation threshold of 1.5 V. Median peak power had fallen to 54% of baseline values by a stimulation intensity of 3.0 V.
Conclusion The findings suggest that DBS can suppress pathological 11–30 Hz activity in the vicinity of stimulation in patients with Parkinson's disease. This suppression occurs at stimulation voltages that are clinically effective.
- Parkinson's disease
- basal ganglia
- deep brain stimulation
- oscillations
- neurophysiology
- electrical stimulation
- motor physiology
- movement disorders
- motor
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Parkinson's disease
- basal ganglia
- deep brain stimulation
- oscillations
- neurophysiology
- electrical stimulation
- motor physiology
- movement disorders
- motor
Introduction
Although pharmacological treatment in Parkinson's disease is initially satisfactory, many patients suffer from severe fluctuations in their clinical state, involuntary movements and prolonged periods of bradykinesia and rigidity after a few years. These problems are difficult to manage and have led to a renaissance of invasive treatment strategies for late-stage Parkinson's disease, particularly deep brain stimulation (DBS) of the subthalamic nucleus (STN). Many potential mechanisms of action of DBS in Parkinson's disease have been suggested.1 One possibility, explored further here, is that DBS suppresses or over-rides pathologically synchronised oscillatory activity which acts as a noisy disruptive signal.2–5 One type of activity in particular has received attention in recordings from patients with Parkinson's disease and involves exaggerated synchronisation at about 20 Hz, in the β frequency band. This is evident in the cross-correlation of neuronal discharges, oscillations of the local field potential (LFP) and spike-triggered averages of LFP activity.3–6
The evidence that DBS may modulate β activity in patients with Parkinson's disease is mixed and mostly indirect. The latter is because simultaneous recordings of STN LFPs during DBS were, until recently, obviated by stimulation-induced electrical artefacts which are several orders of magnitude larger than the spontaneous fluctuations of the LFP.7 Thus, investigators have either recorded at projection sites of the STN, where stimulation artefact is less of a problem, or recorded the immediate after-effects of STN DBS in those patients with a delayed return of bradykinesia upon cessation of DBS. The findings have been mixed, with most studies reporting suppression of β activity,8–12 but one study failing to find such an effect.13 The authors of the latter study have since also recorded from the STN directly during DBS and again failed to show significant suppression of LFP power in the β band.14 Although this study was a technological feat, not all recordings had peaks in the β band prior to DBS so that power suppression may have been difficult to detect in these cases, a problem compounded by the recording of four patients on medication and four patients withdrawn from levodopa. Here, we use similar methodology to study the effects of STN DBS in a larger sample of 16 patients with evidence of pathological synchrony in the subthalamic region at baseline, prior to stimulation. Our aim was to address whether STN DBS suppresses local β activity when this is present.
Methods
A complete description of the methods is available as supplementary material.
Patients and surgery
Sixteen patients participated with informed written consent and the permission of the local ethics committees, and in compliance with national legislation and the Declaration of Helsinki. All had advanced idiopathic Parkinson's disease. Implantation of bilateral STN DBS electrodes was performed sequentially in the same operative session under local anaesthesia, as previously described,15 in all but one patient (case 10; see table in supplementary material). The patients reported here are distinct from those reported by Kühn et al.10
Recordings
Recordings were performed in the few days between electrode implantation and their connection to the pulse generator. The Medtronic electrodes used have four equally spaced contacts. Contact 0 was the lowermost and contact 3 was the uppermost (see supplementary material). Out of the 16 patients, recordings were possible on 28 sides (see table in supplementary material). We used a single-channel, isolated, high-gain (100 dB) amplifier7 with pass band (4–40 Hz) to record LFP signals from the contacts of an electrode while another contact of the same electrode was stimulated. All patients were recorded while they sat in a comfortable chair after overnight withdrawal of their usual antiparkinsonian medication. Patients were instructed to rest quietly, and absence of voluntary movement was confirmed by continuous visual inspection. Initially, about 100 s was recorded from contacts 1/3 and 0/2 on each side with the patient at rest and with no stimulation. The spectral pattern was then analysed immediately off-line in Spike 2 using spectral averages and time-evolving spectral displays. Three of the 28 sides had no discrete spectral peaks, regardless of frequency (see table in supplementary material), and were not studied further. In the remaining 25 sides, we selected the contact pair with the highest peak power (contact pair 1/3 on 19 sides and contact pair 0/2 on six sides) for recording during subsequent stimulation. This was done to maximise our chances of detecting power suppression during DBS (ie, of avoiding a floor effect). The contact pair with the highest β activity has also been previously documented to be well positioned in STN and to concur with the site selected for chronic therapeutic stimulation.15–19
Thereafter, patients were recorded at the selected contact pair on a given side for a further 2 min without stimulation, the latter having been discontinued at least 20 min earlier. Then, unilateral DBS was begun at 1.0 V (19 sides), 1.5 V (five sides) or 2.0 V (one side), depending on time constraints and prior clinical information regarding efficacy. Stimulation was subsequently increased in step increments of 0.5 V (or 1.0 V on two sides) up to 3.5 V, or until side effects were encountered. Each new voltage was maintained for ∼100 s. When time allowed (12 sides in eight patients) at the end of the above slow ramping of stimulation, the latter was then discontinued for 100 s and thereafter stimulation re-presented for a further 100 s at a clinically effective voltage. This was performed so that we could determine whether any suppression of LFP activity during voltage ramping depended on a certain duration of stimulation or on a certain threshold intensity (see results).
Monopolar simulation was delivered by a Medtronic external stimulator (type 3625) between active contacts 1 (when recording from 0/2) or 2 (when recording from 1/3) and a subclavicular surface electrode (pulse-width 60 μs; frequency 130 Hz). Clinical assessment, other than visual inspection, was not made until after all recordings were completed so as to avoid any influence on the LFP. Clinical assessments (items 20, 22 and 24 of UPDRS III) were not blinded. The lowest voltage (threshold) for clinically effective stimulation was determined separately on each side contralateral to the stimulation (see supplementary methods in supplementary material for further details).
Analysis
Spectral analysis was performed in Spike2 v6, using serial FFT blocks of 256 data points (frequency resolution 0.78 Hz, Hanning window, windows not overlapped). These were visualised as time-averaged power spectra (figures 1, 2A) and time-frequency plots (figure 2B). Peaks were defined as local elevations of power in which the five contiguous bins centred on the peak had to be significantly different (p<0.05) to the mean of the two adjacent bins below and three adjacent bins above. One or more discrete peaks were seen in the power spectra from 25 sides. These tended to vary in frequency (5–29 Hz; see table in supplementary material). For this reason, we analysed the amplitude of the LFP in each peak rather than the sum of the LFP power over the whole frequency band of interest. Nineteen sides had a discrete spectral peak in the β 11–30 Hz band, and these were selected for further analysis.

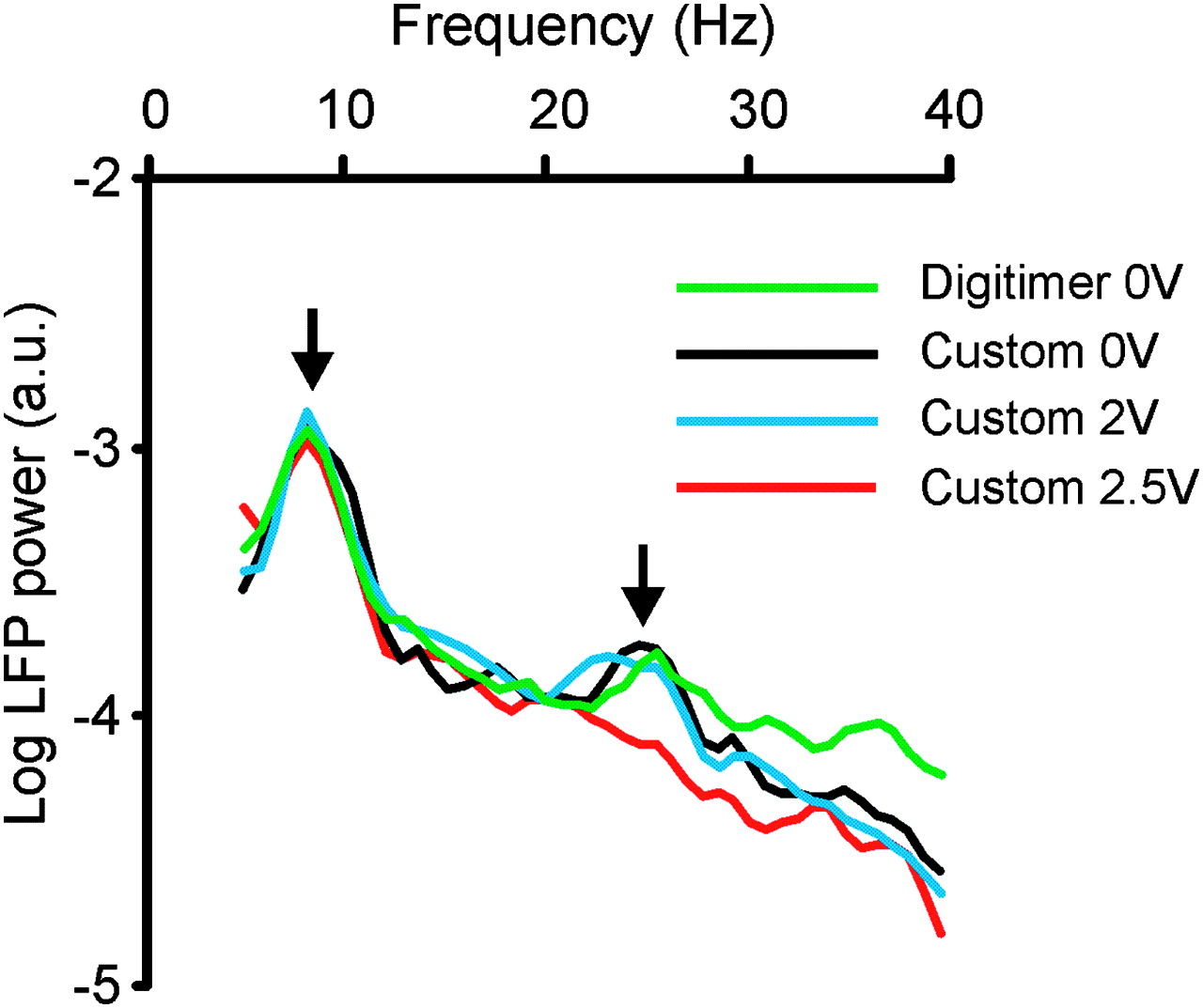
Power autospectra of local field potential (LFP) recorded between contacts 1 and 3 from left subthalamic nucleus deep brain stimulation (DBS) electrode in case 1. Log power autospectra show a peak centred at 8.6 Hz (left vertical arrow), which is unaffected by amplifier type or DBS applied to contact 2 (pulse-width 60 μs; frequency 130 Hz), and a peak centred around 25 Hz (right vertical arrow), which is unaffected by amplifier type but which is suppressed by DBS applied to contact 2 at 2.5 V (130 Hz). The latter was also the threshold for the suppression of contralateral rigidity and bradykinesia as determined by clinical examination using stimulation of the same contact pair performed at a different time on the same day. Two kinds of amplifier were used: Digitimer D360 and our custom-built amplifier. Frequency resolution is 1 Hz. Autospectra averaged over 110 s recorded during same experimental session, but not simultaneously.

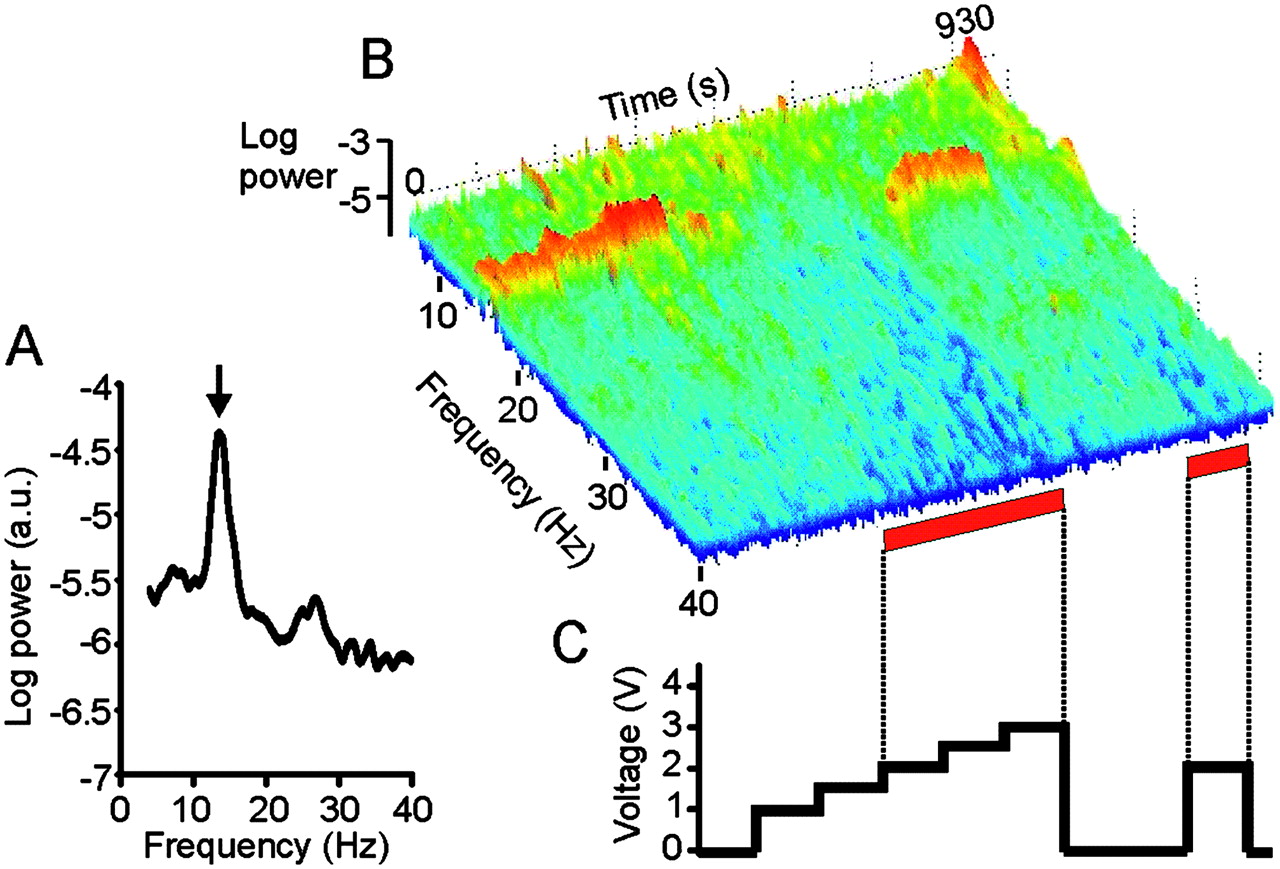
Effect of deep brain stimulation (DBS) of right subthalamic nucleus on local field potential (LFP) in case 5. (A) Power autospectrum of LFP recorded without stimulation. There is a large peak arrowed at 13.6 Hz. (B) Frequency-time log power spectrum of LFP (contact pair 02). Frequency resolution 0.39 Hz. Power, as in (A), shown over the pass band of the amplifier (4–40 Hz). Red bars along the time axis denote periods of DBS at 2.0–3.0 V. Dyskinesias of the contralateral foot were noted at voltages of 2.0 V and above. Note suppression of spectral peak with stimulation ≥2.0 V, with evidence of a temporary increase in the power of the peak with stimulation at 1.5 V and a delayed return of the peak after stimulation at 3.0 V is terminated. (C) Timing and voltage of DBS applied at contact 1. In total, four peaks (on three sides from two patients) between 11 and 30 Hz demonstrated an initial but temporary increase in power before the onset of suppression, as stimulation voltages were progressively increased (see outliers in figure 3).
The beta-band was considered in its entirety and also subdivided into 11–20 Hz and 21–30 Hz bands, following the suggestion that these may have different functional characteristics.20–23 Power in a selected peak was calculated from the sum of the five contiguous bins centred on the peak for each period of stimulation (0–3.5 V). Peak power levels were expressed as percentages of the baseline peak power (in the absence of stimulation). This was performed so as to limit the effects of variability in baseline power between sides, which may, in part, be due to small variations in the positioning of recording contact with respect to the source of the β activity between sides and patients. A similar normalisation procedure is a standard step in the analysis of event related power changes in the EEG, where baseline variability between subjects is also significant. Statistical analyses were performed in SPSS 15 for Windows. As data were not normally distributed, non-parametric statistics were used as detailed in the results (and outliers included). Median values are given, together with the 25th and 75th percentiles (interquartile range or IQ), except when the range was illustrated in a figure. All post-hoc tests specified in the text as p<0.05 survived correction for multiple testing using the false discovery rate procedure.24
Results
The custom-built amplifier performed well so that between 4 and 40 Hz there was little difference between autospectra of LFPs recorded from the subthalamic area with this and commercial devices, provided no DBS was delivered (black and green traces in figure 1). Similarly, there was little difference between autospectra of LFPs recorded with the custom-built device without DBS and with therapeutically subthreshold DBS (black and blue traces in figure 1). However, stronger stimulation voltages suppressed peaks in the 11–30 Hz band (red trace in figure 1). In vitro experiments suggested that DBS induced artefact was unlikely to be responsible for this suppression (see supplementary materials). Note that recordings were made sequentially, rather than simultaneously, which likely accounts for the small differences in the autospectra recorded without DBS in figure 1.
Time-evolving spectra also suggested suppression of LFP peaks between 11 and 30 Hz once a certain threshold voltage of stimulation was surpassed (figure 2). At the group level, there was an effect of stimulation intensity for peaks in the 11–30 Hz band (n=144, χ2=49.1, p<0.00001; Kruskal–Wallis test; figure 3). If the latter band was subdivided into lower and upper ranges, there was also an effect of stimulation intensity for peaks in the 11–20 Hz (n=86, χ2=35.1, p<0.00001) and 21–30 Hz bands (n=58, χ2=15.4, p=0.017; Kruskal–Wallis tests).

{kind=link}
{kind=link}
{kind=link}
Summary box plot of power suppression of beta band peaks during stimulation at different voltages. Power is expressed as a percentage from baseline (without deep brain stimulation). There is a progressive suppression of power of spectral peaks of local field potential between 11 and 30 Hz with increasing voltages. The bottom and top of the boxes are the 25th and 75th percentile, and the band near the middle of the box is the 50th percentile (the median). The whiskers represent one SD above and below the mean of the data. Interrupted lines link medians. Shaded boxes are different from 100% (Mann–Whitney U tests p<0.05, corrected for multiple testing). Peaks with outlying power levels at a given stimulation voltage are denoted by asterisks. The outliers above 150% at 1.5 and 2.0 V are those sides, like those illustrated in figure 2, in which there was a temporary increase in the power of the peak as stimulation voltage was incrementally increased. Peak power suppressed thereafter as stimulation voltage was increased still further.
Post-hoc Mann–Whitney U tests revealed that stimulation at ≥1.5 V suppressed the power in peaks over the 11–30 Hz band (figure 3) and at ≥2.0 V for its subdivisions when these were considered separately (all p<0.05). This threshold compared favourably with the median threshold for the clinical effect of stimulation, determined later using the same contact for stimulation (2.0 V, range 1.5–3.5 V). Moreover, the voltage threshold for peak suppression on individual sides was always 0.5 V less than or equal to the threshold for inducing contralateral foot dyskinesias (case 5 R STN 2 V and case 16L STN 2 V) or tremor suppression (case 2 R STN 2.5 V and case 4 R STN 2.5 V and L STN 3.0 V), where these were evident during the initial simultaneous stimulation and recording session.
With stimulation intensities of 1.0 V, 1.5 V, 2.0 V, 2.5 V, 3.0 V and 3.5 V, the median power of peaks in the 11–30 Hz band fell to 97.5%, 90.5%, 69.6%, 59.6%, 54.2% and 40.5% of baseline values (see figure 3 for ranges and significance levels). Note that this graded suppression in the averaged data partly arose because individual thresholds for β suppression varied. Indeed, four sides showed a temporary increase in peak power at lower stimulation voltages, before peak power was suppressed, as voltage was increased still further (figures 2, 3).
The general picture described above did not change if the frequency band of interest was defined slightly differently as 13–30 Hz, if a broader area of 13 (rather than 5) bins centred on each peak in the β band was analysed or if the two sides in which stimulation-induced contralateral dyskinesias were excluded (see supplemental results). Moreover, similar results were obtained if we only analysed those sides in which stimulation was performed at both 1.5 V and 2.0 V (regardless of any other voltages tested), thereby allowing analysis using the Friedman test for related samples (see supplemental results).
Finally, we addressed a potential ambiguity in the interpretation of the increasing suppression of LFP peaks in the 11–30 Hz band with increasing voltage. As presented above, the increasing suppression could be a direct consequence of the voltage being stepped up or could arise form the progressively longer duration of stimulation. Accordingly, on 12 sides, stimulation was discontinued for 100 s at the end of each series of voltage steps to allow spontaneous activity to recover and then a single clinically effective voltage step re-presented (figure 2). These single steps of higher voltage (median 2.5 V, IQ range 2.0–3.0 V) remained effective at suppressing peaks between 11 and 30 Hz, even though they were not preceded by any prolonged ramping of stimulation (median suppression to 45% of preceding 50 s without stimulation, IQ range 26–97%; z=2.48, p=0.013, Wilcoxon signed ranks test). However, the level of suppression was still less than that observed with the same stimulation intensity after the progressive stepping up of voltages on the same sides (median 35%, IQ range 25–63%; z=2.229, p=0.026, Wilcoxon signed ranks test), suggesting that the duration of stimulation as well as the voltage also had some effect. Relevant in this regard is the finding that longer STN stimulation durations produce longer-lasting after-effects.8
Discussion
We have shown that DBS of the STN area at frequencies and voltages in the therapeutic range leads to a concurrent suppression of locally synchronised activity between 11 and 30 Hz, where the latter is evidenced by a peak in baseline spectra of LFP activity over this frequency range. Such β activity is believed to be pathological4 and is much attenuated in healthy animals, and, in implanted dystonic patients, is only seen in those made Parkinsonian by the use of chronic tetrabenazine.25 Thus, the present study bears out the assumption that the suppression of pathological oscillatory activity seen temporarily after cessation of DBS is a reflection of suppression during DBS.8 10 12 The LFP is currently believed to mainly reflect slow subthreshold currents, primarily postsynaptic potentials, of a large local neuronal population26 and is considered to reflect mainly the ‘input’ to the local network.27 This interpretation is supported in the case of the β band LFP activity recorded in the STN by the fact that it is coherent with, but lags, similar oscillatory activity in the cerebral cortex, in line with cortical driving.22 28 29 Viewed in this light, our results are compatible with the recent demonstration in a rodent model that it is the afferents to the subthalamic nucleus that must be stimulated at high frequency, rather than the local neurons themselves, to overcome parkinsonism.30
The above rodent study also showed that stimulation of cortical afferents at 20 Hz greatly exacerbated parkinsonism.30 Indeed, synchronisation in the β band has been implicated in parkinsonian bradykinesia and rigidity in both correlative and interventional studies,3–6 so its suppression by subthalamic stimulation using standard therapeutically effective parameters raises the possibility that this suppression of local β activity may underlie some of the therapeutic actions of DBS. This is not to say that the effects of this suppression are limited to the STN region, or that this is the sole consequence of stimulation. It is possible that DBS influences neuronal activity both locally at the site of stimulation, that is in and around STN, and over other functionally connected elements of the cortex–basal ganglia network.1 9 10 31 32 This could arise either directly through the suppression of synchronised activity at the stimulation site, so that this activity is no longer propagated, or through the effects of stimulation-induced high-frequency activity of STN neurons and axons in the vicinity.1 32 33 The latter would not have been detected by our amplifier, which necessarily had a frequency cut-off well below this. Any induced high-frequency activity might then be transmitted to the output structures of the basal ganglia34–36 where it might have further effects, including suppression of pathological activity at these and later relay stages. Additionally, stimulation may lead to antidromic spikes that collide with ongoing spontaneous pathological activity, such as might come from the cortex.1 This could contribute to the suppression of synchronisation in the subthalamic region, given that much of this may be driven by the cerebral cortex.22 28 29
Our study was constructed to test whether deep brain stimulation can suppress local β activity when this is present. We argued that there was little point proceeding to stimulation if there was no peak at rest that could potentially be suppressed. Possible reasons for an absent β peak at rest include suboptimal targeting,15 stun effect15 36–38 and incomplete withdrawal of antiparkinsonian medications (through either the long-lasting nature of some dopamine agonists or covert levodopa ingestion). Still, those few cases in which spectral peaks are absent in the β band during the postoperative period raise the possibility that DBS-induced suppression of such activity may not explain clinical improvement in every patient. In line with this, although suppression of β activity correlates with treatment-induced improvement in bradykinesia and rigidity, it does not correlate with improvement in parkinsonian tremor, so that even in patients with clear β activity, this may not relate to all motor impairments.39–41 However, it is also possible that pathological synchrony may, in the minority of our cases, have predominated at sites in the basal ganglia–cortical circuit other than the STN. We only measured synchronisation in the subthalamic region and yet, as discussed above, high-frequency stimulation of the STN area may have effects at several sites in the distributed basal ganglia–cortical circuit.
Although the present data indicate that DBS of the STN at clinically effective voltages is associated with the simultaneous suppression of oscillatory activity in the β band, they do not address the question of the precise site of origin of such LFP activity. In particular, whether β activity is confined to the dorsal STN or extends for a few millimetres above its dorsal border is unclear. Support has been provided for both possibilities, depending on whether LFPs18 42 or population spiking activity43 are studied. Both DBS of the dorsal STN and caudal zona incerta have clear antiparkinsonian effects.44–46
The present study suffers from several limitations. In order to recruit a large patient sample, we studied patients at several different surgical centres, which may have introduced additional variance in our data set. Another limitation was the absence of kinematic assessment that might have allowed us to compare the threshold stimulation voltage for significant power suppression with the voltage that objectively improved motor performance in all patients. This issue should be addressed in future studies. Nevertheless, our results demonstrate a potentially important association between the suppression of local oscillatory synchrony and DBS in the therapeutic range.47 Note, that there was no significant difference in the DBS-induced suppression of peak power in the lower and upper frequency bands, when the β band was split for analysis. This indicates that although the functional significance of the subdivisions within the β band remains debated, they are both suppressed by STN DBS.
The present study provides direct evidence that STN DBS can reduce β frequency activity in the region of the STN. It is interesting to note that levodopa has a similar effect,16 38–40 48 49 raising the possibility that suppression of such oscillatory synchrony by several treatment modalities may contribute to the amelioration of some aspects of parkinsonism. Moreover, even if the suppression of β activity were found to be epiphenomenal rather than causally important in parkinsonism, the present results are important in reinforcing the suitability of subthalamic LFP activity in the β band as a feedback signal for closed-loop DBS systems.50 This activity correlates with bradykinesia and rigidity38–40 51 and is suppressed by DBS.
Acknowledgments
We thank P Limousin and M I Hariz from the Unit of Functional Neurosurgery (Institute of Neurology, 33 Queen Square London WCIN 3BG) for giving us permission to study their patients.
References
Footnotes
P Brown holds a consultancy with Medtronic Inc.
Funding This work was supported by the Medical Research Council, Wellcome Trust, the Oxford NIHR Biomedical Research Centre, Parkinson's Disease UK and Fondation pour la Recherche Médicale.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the Joint Ethics Committee of the National Hospital for Neurology and the Institute of Neurology.
Provenance and peer review Not commissioned; externally peer reviewed.