Article Text

Abstract
Background Relatively few studies have searched for potentially pathogenic antibodies in non-paraneoplastic patients with cerebellar ataxia.
Methods and Results We first screened sera from 52 idiopathic ataxia patients for binding of serum IgG antibodies to cerebellar neurons. One strong-binding serum was selected for immunoprecipitation and mass spectrometry, which resulted in the identification of contactin-associated protein 2 (CASPR2) as a major antigen. CASPR2 antibodies were then found by a cell-based assay in 9/88 (10%) ataxia patients, compared to 3/144 (2%) multiple sclerosis or dementia controls (p=0.011). CASPR2 is strongly expressed in the cerebellum, only partly in association with voltage-gated potassium channels.
Conclusions Prospective studies are now needed to see whether identification of CASPR2 antibodies has relevance for the diagnosis and treatment of idiopathic cerebellar ataxia.
- CASPR2
- autoimmune
- cerebellar ataxia
- VGKC-complex
- antibody
- neuroimmunology
- neurooncology
- NMDA
- paraneoplastic syndrome
- stiff man syndrome
- biochemistry
- molecular biology
- multiple sclerosis
- neuroepidemiology
- neurogenetics
- myasthenia
- channels
- lambert eaton syndrome
- stiff man syndr
- limbic system
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- CASPR2
- autoimmune
- cerebellar ataxia
- VGKC-complex
- antibody
- neuroimmunology
- neurooncology
- NMDA
- paraneoplastic syndrome
- stiff man syndrome
- biochemistry
- molecular biology
- multiple sclerosis
- neuroepidemiology
- neurogenetics
- myasthenia
- channels
- lambert eaton syndrome
- stiff man syndr
- limbic system
Introduction
Cerebellar ataxia is a relatively common syndrome with diverse causes. Some patients have a paraneoplastic aetiology associated with autoantibodies to intracellular antigens, such as Yo (PCA-1), but these antibodies are unlikely to be directly pathogenic, and the patients seldom respond well to immunotherapies. In the last few years, antibodies to neuronal surface antigens have been demonstrated in patients with immunotherapy-responsive forms of limbic encephalitis and related disorders,1 2 raising the possibility that other CNS disorders may also result from autoantibodies to cell-surface proteins. There have been some previous reports of potentially pathogenic antibodies in cerebellar ataxia, such as voltage-gated calcium channel (VGCC) antibodies,3 glutamic acid decarboxylase (GAD) antibodies mainly in patients with polyendocrine syndromes,4 a small number of patients with mGluR1 antibodies,5 and associations of cerebellar ataxia with gluten sensitivity and gliadin antibodies,6 but there have been few systematic cohort studies to identify new antigens. Here we have identified a potentially pathogenic antibody against the neuronal membrane protein contactin-associated protein 2 (CASPR2) in nine of 88 (10%) patients with unexplained ataxia.
Subjects and methods
Clinical material
Twenty-five Spanish sera (Valencia, 10; Barcelona, 15) were from patients with acute or subacute cerebellar ataxia (<3 months duration at testing) referred for onconeural antibody testing, with some evidence of an autoimmune mechanism (CSF raised cells or oligoclonal bands, partial response to immunotherapy and/or spontaneous remission) but with no serological or imaging evidence of tumours, and exclusion of other causes including infectious disorders. We also studied 27 and subsequently a further 36 sera from Welsh patients with idiopathic late onset cerebellar ataxia who had been recruited to a population-based study in south Wales between 1999 and 2008 and in whom known causes or associations (eg, coeliac disease) had been excluded.7 All three centres had ethics approval for the study of these patients' sera. Control sera included 101 from patients with multiple sclerosis and 43 from patients with dementia. Radioimmunoprecipitation assays were used to look for antibodies to VGCC, GAD and voltage-gated potassium channel complexes (VGKC-complex) as previously described.3 4 8
Antibody-binding assays
Cerebellar organotypic slice cultures were prepared from 9-day-old mice and dissociated cultures of cerebellar granule neurons (CGNs) were prepared from 5-day-old mice as previously described.9 10 Antibody-binding assays were performed on organotypic slices after 12 days (P9+DIV12) in culture and on CGNs after 10 days (P5+DIV10). Unfixed slices or neurons were incubated with patient sera (1:125) in serum-free culture media supplemented with 25 mM Hepes and 1% bovine serum albumin for 1 h at room temperature (RT), washed three times and fixed with 3% formaldehyde in phosphate-buffered saline for 30 min (slices) or 15 min (CGNs) at RT. Subsequently, slices were permeabilised with methanol for 5 min at −20°C. After three washes, slices and CGNs were incubated with anti-human IgG Alexa Fluor 568-conjugated secondary antibody (Invitrogen, Carlsbad, California, USA) for 45 min at RT. Slices were counterstained with an anti-calbindin antibody (Swant, Marly, Switzerland) to label Purkinje neurons. Slices and CGNs were washed and mounted with mounting medium containing DAPI (Vectashield; Vector Laboratories, Burlingame, California, USA).
The cell-based assay (CBA) was performed as described.8 Briefly, human embryonic kidney cells (HEK293T) (American Type Culture Collection) were transfected with EGFP-tagged CASPR2. Then, 48 h post-transfection, live cells were incubated with patient sera initially at 1:50 and subsequently at 1:100 for 1 h at RT and then fixed and stained as described above. Antibody binding at 1:100 was visualised using an immunofluorescence microscope and scored by at least two independent blinded observers from 0 to 4 as in previous studies.11 12 The final score was the median of scores of two to three independent assays for each serum (variance <1).
Immunoprecipitation and mass spectrometry
Live CGNs in culture (P5+DIV10) were incubated with patient sera (1:40) in serum-free culture media buffered with 25 mM Hepes for 30 min at RT. Subsequently, neurons were washed twice and solubilised with 10 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% TritonX-100 and protease inhibitors for 45 min rotating at 4°C. Samples were centrifuged for 15 min at maximum speed at 4°C, and the supernatants were incubated with protein A-agarose for 5 h at 4°C. The beads were washed five times and resuspended in SDS sample buffer, boiled and analysed by gradient SDS-PAGE (Nupage; Invitrogen) with Coomassie staining (Imperial Protein Stain; Thermo Scientific, Rockford, Illinois, USA). Coomassie-stained bands were extracted as previously described.13 Digested protein material was analysed by liquid chromatography using an Ultimate 3000 nano-HPLC system interfaced with a 3D high capacity ion trap mass spectrometer (AmaZon; Bruker Daltonics, Billerica, Massachusetts, USA). Raw LC-MS/MS data were processed using DataAnalysis 3.4 (Bruker Daltonics) and Mascot software with the SwissProt database.
Results
To identify surface antigens that might mediate autoimmune cerebellar ataxia, we first screened the 25 Spanish and 27 of the Welsh sera. None had onconeural antibodies, evidence of a tumour, or an identifiable genetic or other cause. One patient had very high antibodies to GAD (4000 U/ml), four patients had low levels of antibodies against VGCC (range 84–122 pM, normal values (nv) <45 pM), and two patients had antibodies to VGKC-complexes (757 and 165 pM, nv <100 pM).
We tested the 52 sera for IgG binding to live, unpermeabilised cerebellar neurons using organotypic cerebellar slice cultures and dissociated primary CGNs. We identified 14 sera that showed positive staining on cerebellar neurons and no significant binding with seven healthy sera, all observed in a blinded manner. We focused on the serum that bound most strongly in a punctate manner to the axons of the CGNs in the molecular layer of the slices and to dissociated CGNs (figure 1A,B). To identify the neuronal surface proteins recognised by the patient serum IgG, we performed an immunoprecipitation from the granule neuron cultures using this patient's and one healthy control serum. A 180-kDa polyprotein was selectively precipitated by the patient serum (figure 1C) and identified as contactin-associated protein like-2 (CASPR2) by mass spectroscopy. The other distinctive bands yielded no significant hits (data not shown).
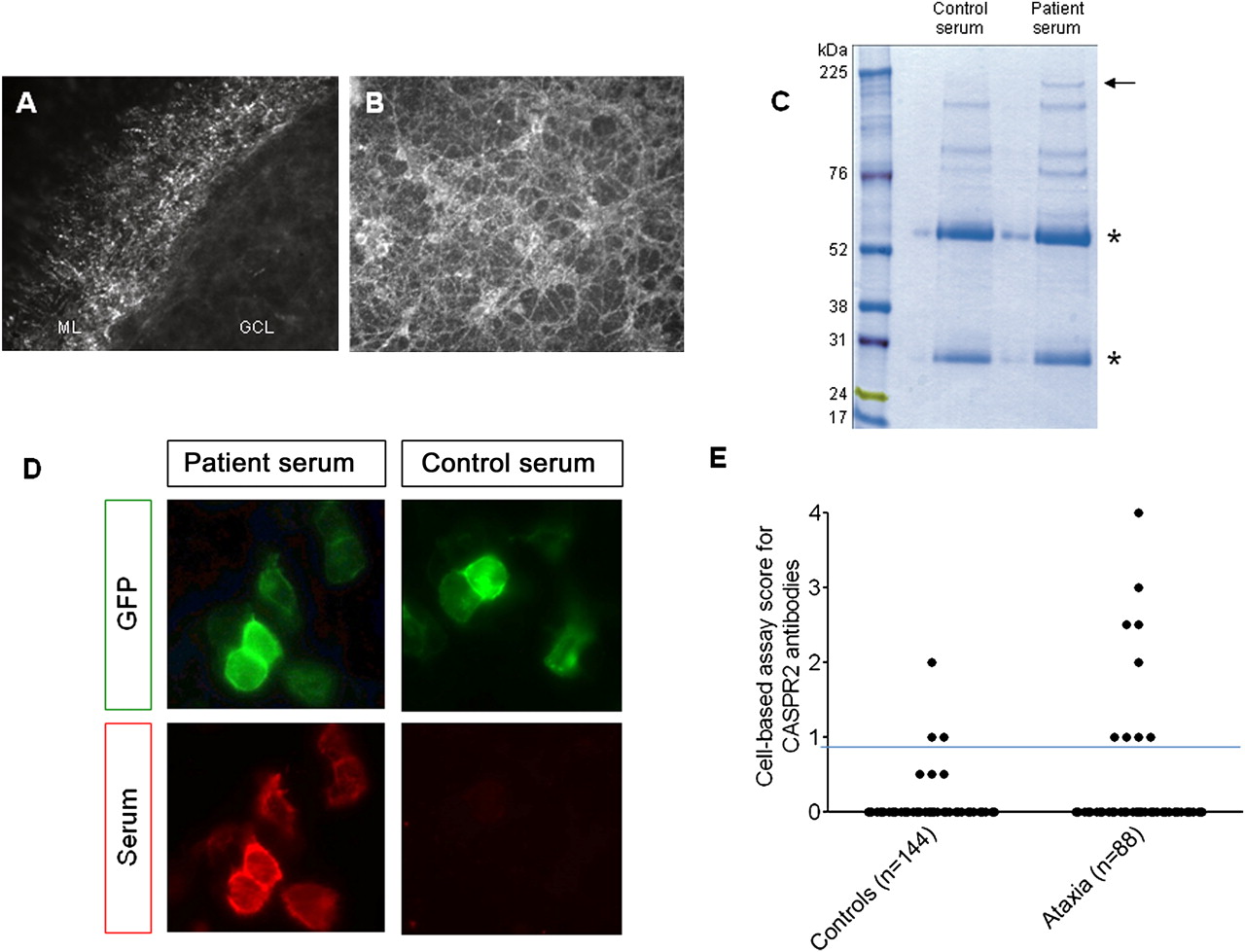
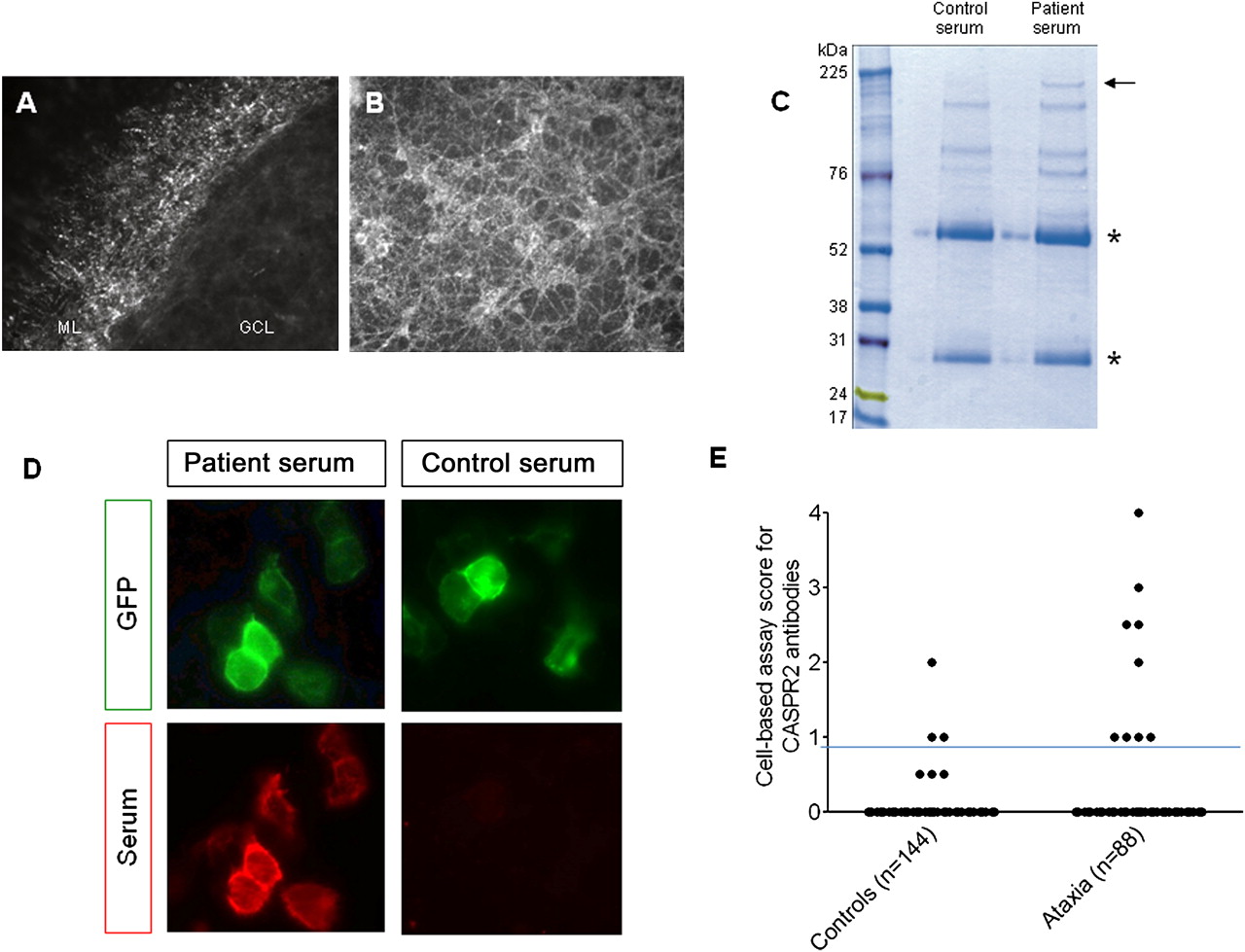{kind=link}
The serum bound strongly, in a punctuate manner, to the axons of cerebellar granule neurons (CGNs) in the molecular layer of unpermeabilised organotypic cerebellar slices (A) and unpermeabilised dissociated CGN cultures (B). GCL, granule cell layer; ML, molecular layer. (C) Gel electrophoresis of immunoprecipitates after incubation of serum with unpermeabilised CGNs followed by solubilisation. The asterisks indicate heavy and light IgG chains, respectively. All precipitated proteins were excised and analysed by mass spectrometry. CASPR2, corresponding to the 180-kDa protein band selectively precipitated by the patient's IgG (arrow), was the only significant membrane protein identified by mass spectrometry (MudPIT scores >41). (D) The index patient serum IgG (red) bound strongly to the CASPR2-EGFP-transfected (green) HEK293T cells (score 4) but the control serum did not bind (score 0). (E) Binding in the cell-based assay was determined at 1:100 serum dilution and scored visually from 0 (no binding) to 4 (very strong binding). Sera scoring 1 or above were considered positive, as in our previous publications, and only if scoring 1 or above in two independent, coded assays. Positive binding to CASPR2 (score 1–4) was detected in nine of 88 (10%) patient sera compared with three of 144 control samples (Fisher's exact test, p=0.011). The horizontal line represents the cut-off above which the results are considered positive. To confirm some of the lower values, available sera were tested to endpoint dilution. Two MS sera scoring 1 and 2 titrated to 1:100 and 1:200; two ataxia sera scoring 1 and 1.5 titrated to 1:200 and 1:400.
To confirm the presence of CASPR2 antibodies in this patient's serum, and to test the whole cohort, we employed a cell-based assay using full-length GFP-tagged human CASPR2 expressed in HEK293T cells.8 We scored the binding of the serum antibodies with a visual scoring system on coded samples that has been well-validated for other antibodies11 12 and is used for routine diagnosis. The index patient serum bound strongly to the CASPR2 cells (score 4; normal values <1; figure 1D), but not to untransfected cells or control-transfected cells (not shown). An additional six sera (three of those positive on slices) also bound to CASPR2, with scores between 1 and 3 (total 7/52). We then tested another 36 ataxia patients and found a further two CASPR2-antibody positive patients, making a total of 9/88 (10%); one additional patient had VGKC-complex antibodies by immunoprecipitation (186 pM) but none were positive for GAD antibodies. Only 3/144 (2%) disease control sera tested in parallel were positive for CASPR2 antibodies (scores 1–2). All results are shown in figure 1E.
Information on the CASPR2-antibody positive patients is given in table 1. Most had a subacute or insidious onset mainly with a progressive course, but one with the lowest CASPR2 antibody had an acute onset, fully reversible cerebellar ataxia. Six of the patient samples were not taken early in the course of the disease. The index patient, who had the high VGKC-complex antibody titre of 757 pM, was a man of 59 years of age whose predominant presenting features were slurred speech, unstable gait and personality changes, followed by a nocturnal generalised tonic-clonic seizure. MRI, CSF and EMG were reported as normal. Although the patient was a heavy smoker, all tumour screens were negative. He progressively deteriorated with difficulties in phonation, swallowing and walking, as well as psychiatric disturbance, and probable temporal lobe seizures. CSF was normal and viral screen negative. By that time, there were mild FLAIR hyperintensities in both hippocampi, with mild cerebellar atrophy, and interictal EEG showed bilateral temporal epileptiform discharges. VGKC-complex antibodies were first found to be positive at this stage, and the patient was treated with high dose intravenous immunoglobulins with partial temporary amelioration of symptoms including the ataxia; cyclosporine was introduced a year later and used for 3 years. When last seen, the patient was stable on lamotrigine and sertraline only, with scanning dysarthria and mild ataxia but able to walk in open spaces. He had monthly temporal partial seizures but his mood, behaviour and memory were normal. All the patients were negative for LGI1 antibodies (data not shown).
Summary of clinical and serological data in eight patients with CASPR2 and/or VGKC-complex antibodies
Discussion
We identified CASPR2 as an antigenic target in 10% of patients with idiopathic, cerebellar ataxia. CASPR2 is a transmembrane protein with a large extracellular domain and is highly expressed in the axons of the granule neurons of the cerebellum,14 thus representing an excellent candidate antigen for cerebellar ataxia. CASPR2 is essential for the clustering of the VGKC subunits Kv1.1 and Kv1.2 at juxtaparanodal regions of myelinated axons and at the axon hillock15 and recently has been described as one of the components of the VGKC-complex that is the target for antibodies in patients with limbic encephalitis, Morvan's syndrome and acquired neuromyotonia.8 16 However, in this study, only one of nine CASPR2-antibody positive patients was positive by radioimmunoprecipitation for VGKC-complex antibodies. This patient presented with ataxia and also had some limbic involvement, but none of the other CASPR2-antibody positive patients had evidence of limbic encephalitis. The lack of VGKC-complex antibodies in the remaining patients suggests that CASPR2 antibodies in ataxia may recognise CASPR2 epitopes that are independent of the VGKC-complex. Indeed, immunostaining of rodent cerebellar tissue shows strong CASPR2 staining in the cerebellar granule and molecular layers but not in the strongly Kv1.1-positive cerebellar pinceau.8 Thus, CASPR2 may have additional functions in the cerebellum as well as its established role in juxtaparanodal VGKC clustering, and the antibodies may bind preferentially to these CASPR2s.
Whether CASPR2 antibodies have a primary pathogenic role in ataxia, and their mechanisms of action, remain to be demonstrated. Although three of nine patients had recent onset of disease, and one of these had full recovery, six patients had a longer history at sampling; in these patients it is possible that CASPR2 antibodies represent a secondary phenomenon. However, lower levels of CASPR2 antibodies might represent the temporal tail of a higher antibody response present initially, since many of the newly identified neurological diseases associated with antibodies to neuron surface proteins are monophasic.1 2 There were patients studied here whose serum IgG bound to cerebellar slices or cultured CGNs but did not bind to CASPR2 or to VGKC-complexes, VGCCs or GAD. Although we did not attempt to perform mass spectrometry with these patients' sera, it is likely that other antigenic targets could be identified in the future. It is therefore important that future serological studies examine recent-onset patients and look for antibodies to CASPR2 and other novel antigenic targets. Immunotherapies could then be applied to those patients in whom a potentially pathogenic antibody is found, in the hope that some improvement or stabilisation will occur before permanent cerebellar damage.
Acknowledgments
We are grateful to Drs J Palace, KP Wandinger and R Nilforooshan for use of the control sera, and thank Professor K Davies for support.
References
Footnotes
Funding EB was funded by an OXION Wellcome Trust fellowship, LZ was a recipient of an EFNS fellowship, and PW and AV are supported by the NIHR Oxford Biomedical Research Centre.
Competing interests AV and the University of Oxford receive royalties and payments for antibody assays. AV holds a patent for VGKC-complex antigens for the detection of antibodies. AV, PW and BL may receive royalties resulting from licensing this patent.
Ethics approval The ethics committees in Cardiff, Barcelona and Valencia approved this study.
Provenance and peer review Not commissioned; externally peer reviewed.