Article Text

Statistics from Altmetric.com
Introduction
The term leukodystrophy refers to a group of conditions that are inherited and involve the progressive destruction or loss of previously acquired myelin.1 The most commonly reported of these disorders have a metabolic origin and are associated with abnormalities on specialist biochemical testing. Recently, a number of conditions caused by genes coding for proteins not directly involved in metabolic pathways and for which the diagnosis relies directly on gene analysis have also been described. In clinical practice, distinguishing ‘classical’ inherited leukodystrophies from other causes of white matter disease, including vascular and inflammatory disorders, may not always be straightforward.
Although individually rare, with no single condition having a prevalence of >1 in 20 000, the reported prevalence of adult onset leukodystrophies is rising. This is likely to be related to the increased use of brain MRI and new genetic insights. Collectively their incidence may rival that of multiple sclerosis (MS).2 Nonetheless, the rarity of each condition and the wide differential means that diagnosis can be challenging and most clinicians will lack experience in the area. Currently, a significant proportion of individuals may remain without a precise diagnosis despite intensive investigations.
Much has been written in the paediatric literature about leukodystrophies,3 but the adult neurologist, with a new case, is often left with an extensive and detailed table of rare disorders to consider, without an obvious diagnostic pathway to follow. In addition, leukodystrophies that classically present in infancy or childhood may have a very different or attenuated clinical presentation in adulthood, making diagnostic features less familiar. In this review, taking a clinical case as a starting point, we address:
-
▸ the most commonly presenting leukodystrophies in adults
-
▸ their clinical presentations
-
▸ MRI patterns for specific leukodystrophies
-
▸ how to investigate leukodystrophies in an adult
-
▸ current and future treatment possibilities.
Table 1 summarises the most frequent of the inherited leukodystrophies that have been reported to present in adulthood (age of onset of >16 years). This list is not exhaustive and almost certainly underestimates the true numbers—with more widespread access to genetic testing it is likely that the list of potential diagnoses and actual prevalences will continue to increase.
Leukodystrophies presenting in adulthood
Case study
A middle aged patient presented to an MS clinic with a 2-year history of progressive gait and cognitive difficulties. Two years earlier, the patient had noted dragging of the right leg, difficulty controlling the leg and had had several falls. One year prior, clumsiness and weakness in the right hand had been noted, as well as word finding difficulties, slowness of thought, problems with episodic memory and that their writing and spelling had deteriorated. The patient had a past history of sciatica and long standing depression. The patient's mother had died at 65 years of age after a 5-year decline in gait and cognitive function, with MRI brain showing generalised cerebral atrophy and patchy signal change in the peri-ventricular white matter. The mother had been given a diagnosis of vascular dementia. There was no other family history of neurological or psychiatric illness.
On examination, the patient had broken smooth pursuit eye movements and hypometric saccades. The cranial nerves were intact. The upper limb examination was normal. The lower limb examination showed increased tone and power of 4/5 on the right, with a pyramidal pattern of weakness. The reflexes were brisk throughout, with equivocal plantar responses. The gait was apraxic. On cognitive testing she scored 22/30 on the Mini-Mental State Examination. On neuropsychometric testing, there was evidence for generalised cognitive decline with marked impairments on tests sensitive to frontal and parietal lobe functions.
MRI brain (see online supplementary figure S1) showed extensive white matter change predominantly affecting the frontal lobes with involvement and thinning of the corpus callosum.
What is the differential diagnosis and which initial investigations should be performed?
With this symptom complex and widespread white matter change on MRI, there are a number of acquired conditions that need to be excluded before an inherited leukodystrophy is considered. These include inflammatory, autoimmune, infectious, neoplastic, metabolic, drug and toxic causes. Suggested initial (Round 1) investigations are outlined in online supplementary table S1. A very rapid onset and progression over months is much more likely to be acquired than genetic, though cerebral adrenoleukodystrophy (ALD) can present abruptly.
Case discussion: Round 1 investigations
In this case, the phenotype was not one of relapsing–remitting MS, but a progressive deterioration, characterised by cognitive involvement over the previous 2 years. While progressive cognitive changes can occur in MS, the rapidity and severity are unusual. There also seems to be a possible dominant or maternal family history, which can occur in MS, but again uncommonly.
The following Round 1 investigations were normal or negative: full blood count, electrolytes, liver function tests; autoimmune and vasculitic screen. CSF examination revealed a mildly elevated protein of 0.58 g/L (normal <0.40 g/L), no white cells and was negative for oligoclonal bands (<5% of cases of MS).38 Visual evoked potentials showed no abnormalities and nerve conduction studies were normal.
What other information may be helpful in directing investigations?
(A) Family history: This, if present, can be very informative (table 1). In establishing the inheritance pattern of a condition, the occurrence of sporadic cases, non-paternity, misdiagnosis in previous generations/other family members and a varied phenotypic presentation of the same disorder within a family should be considered.
(B) Ethnicity: The ethnic background of the patient may be helpful in focusing investigations (table 1). Conditions such as mucolipidosis, adult onset gangliosidosis and adult polyglucosan body disease are more common in the Ashkenazi Jewish population, whereas megalencephalic leukodystrophy with cysts is more common in those of Asian-Indian or Turkish ethnicity.
(C) Clinical presentation: Table 2 details particular clinical presentations that may point to a certain leukodystrophy. Absence of a feature does not however exclude a condition.
Clinical features associated with adult onset leukodystrophies
(D) MRI pattern: Specific changes on brain MRI can have diagnostic utility, but there is considerable overlap among disorders (table 3/figure 1). Table 3 details the MRI findings classically associated with the various leukodystrophies. Figure 1 illustrates the typical site of brain MRI changes for particular conditions. Figures 2 and 3 show brain MRI scans of adult patients with confirmed leukodystrophy. Additional information can be obtained from cervical imaging (eg, spinal cord atrophy seen in adult polyglucosan body disease) as well as cranial imaging and the use of contrast enhancement (eg, Alexander disease). Schiffmann and van der Knapp39 provide a diagnostic algorithm using MRI for the diagnosis of white matter disorders that uses features such as the location of white matter change and the involvement of the peripheral nervous system to aid diagnosis.
Specific MRI findings in leukodystrophies
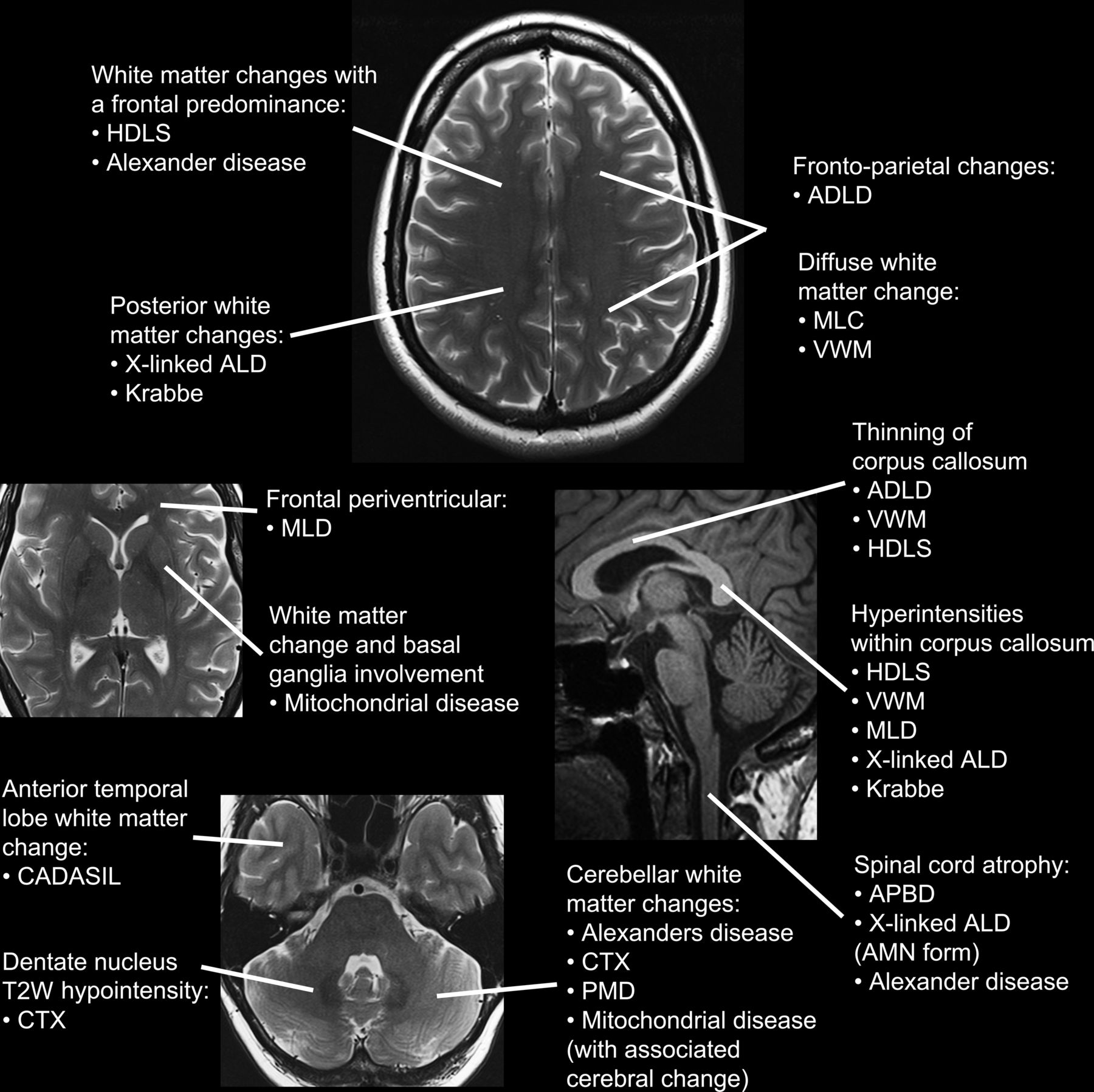
Schematic illustration of characteristic anatomical distribution of MRI abnormalities on normal MRI sequence panels, clockwise from the top: axial T2-weighted section at the level of the centra semiovale, midline sagittal T1-weighted section through the corpus callosum, brainstem and upper cervical spinal cord, axial T2-weighted section through the temporal poles, cerebellum and dentate nuclei and axial T2-weighted section through the basal ganglia.

MRI brain acquisitions of: X linked adrenoleukodystrophy on axial T2-weighted (a), coronal FLAIR (b) and coronal postgadolinium contrast (c) sequences showing diffuse white matter signal T2-weighted hyperintensity and volume loss of the posterior frontal, posterior callosal and parieto-occipital regions, extending along the corticospinal tracts through the internal capsules and temporal periventricular white matter. Peripheral enhancement of the parieto-occiptal confluent symmetrical signal abnormality is demonstrated. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy on axial T2-weighted (d, e) and coronal FLAIR (f) sequences showing diffuse symmetrical white matter hyperintensity extending through the external capsules and temporal poles with multiple mature subcortical and a left superior parietal cortical infarcts. Krabbe disease (globoid cell leukodystrophy) on axial T2-weighted (g) and coronal FLAIR (h, i) sequences showing posterior periventricular and deep white matter hyperintensity and volume loss involving the corticospinal tracts and splenium of the corpus callosum.

{kind=link}
{kind=link}
{kind=link}
MRI brain acquisitions of: cerebrotendinous xanthomatosis on an axial T2-weighted sequence through the posterior fossa (a) demonstrating white matter signal hypointensity involving the dentate nuclei and surrounding hyperintensity. Note the diffuse volume loss of the cerebellum disproportionate to the visible temporal poles. Metachromatic leukodystrophy on an axial T2-weighted sequence through the centra semiovale (b) demonstrating widespread symmetrical white matter signal hyperintensity sparing the subcortical U fibres and radiating transmedullary bands. Mitochondrial encephalopathy lactic acidosis and stroke-like episodes on an axial FLAIR sequence (c) demonstrating swelling and patchy hyperintensity of the cortical and subcortical matter of the occipital, right mesencephalic and left lateral temporal regions during an acute stroke-like episode. Adult polyglucosan body disease on an axial (d, e) and sagittal (f) FLAIR sequences demonstrating diffuse periventricular and inferior callosal white matter hyperintensity and volume loss. There is similarly cerebellar and spinal cord volume loss. An incidental planum sphenoidale meningioma is present. Vanishing white matter disease on axial and sagittal T2-weighted (g, i) as well as coronal T1-weighted (h) sequences demonstrating diffuse white matter T2-weighted hyperintensity and volume loss. Atrophy of the corpus callosum and spinal cord is also present.
Leukodystrophies and other white matter conditions can be misdiagnosed as MS.40 MRI white matter changes need to be examined closely for any ‘red flags’ that would not be consistent with a diagnosis of MS and may point towards another diagnosis.41 These have been suggested by the workshop of the European Magnetic Resonance Network in Multiple Sclerosis41 to alert clinicians based on imaging findings to consider another diagnosis. They include large lesions with mass effect, ring enhancement and symmetrically distributed lesions. Typically, findings in MS include: preferential involvement of the subcortical U fibres, the corpus callosum, temporal lobes, brainstem and cerebellum. Periventricular areas of T2 high signal are seen in 95% of patients, which may be scattered or confluent. Use of the McDonald 2010 criteria for the diagnosis of MS has been estimated to have an accuracy of 86% and specificity of 93%.42 In addition, spinal cord involvement may commonly be seen, unlike most genetic leukodystrophies, underscoring the value of cervical MRI. A rare differential diagnosis for MS is retinal vasculopathy with cerebral leukodystrophy, an autosomal dominant condition associated with pseudotumours on imaging, white matter lesions, visual changes and possible systemic findings.43
A common cause of white matter change is small vessel cerebrovascular disease. Leukodystrophies, as with the mother of this patient, may be diagnosed as vascular dementia. While small vessel ischaemia can cause very significant white matter change, red flags include: extensive changes in the absence of significant vascular risk factors and the absence of basal ganglia or brain stem involvement. Cerebral amyloid angiopathy can also lead to white matter changes; here, T2* or susceptibility weighted imaging can be helpful in demonstrating ‘microbleeds’ which are typically peripherally located. There have been reports of white matter change particularly involving the parieto-occipital regions in patients with duplication of the amyloid precursor protein.44 MRI changes of other differential diagnoses include the simultaneous enhancement of lesions and punctiform parenchymal involvement seen in sarcoidosis; mass effect and ring enhancement seen in abscesses; and bilateral non-progressive lesions at the grey–white matter junction in acute demyelinating encephalomyelitis.
Case discussion: family history, ethnicity, clinical phenotype and MRI pattern
Returning to the case study and considering points (A)–(D) above, of particular note is the history of early onset dementia in the patient's mother, although definitive diagnostic information was lacking and there was no suggestion of other family members having been affected. Often the family history is censored or unclear. In this case, the history is consistent with maternal or dominant inheritance (A). From table 1, the four principal leukodystrophies associated with an autosomal dominant inheritance are: hereditary diffuse leukoencephalopathy with neuroaxonal spheroids (HDLS), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), Alexander disease and adult onset autosomal dominant leukodystrophy (ADLD). The ethnicity provided no additional information (B). Our case had no features of migraine or stroke-like episodes to suggest CADASIL; no bulbar/pseudobulbar involvement or morphological features (eg, macrocephaly) to suggest Alexander disease; and no autonomic involvement to suggest ADLD (C). The MRI (D), which showed widespread white matter change with a frontal predominance and atrophy of the corpus callosum, was most suggestive of HDLS or ADLD. Other leukodystrophies with frontal predominance (table 3) include metachromatic leukodystrophy (MLD); however, this has an autosomal recessive pattern of inheritance.
At this stage, it is appropriate to proceed to more specialist Round 2 investigations (table 4). From table 1 it can be seen that (fasting) very long chain fatty acid (VLCFA) levels, relevant white cell enzyme activities, cholestanol profile, urine bile alcohols and plasma amino acids should be measured as these will screen for the more common metabolic leukodystrophies. In particular, ALD has the highest prevalence at 40/million apart from the combined mitochondrial disorders, and should be tested early. Before testing, it is advisable to contact the local specialist laboratory to discuss the type of sample required, the optimum timing of samples and the storage/transport conditions required to ensure integrity of the sample for testing.
More specialised Round 2 investigations
In order to aid interpretation of the results, the laboratory should be informed of the age of the patient, the age of onset of the symptoms, and a summary of relevant clinical findings and family history. A list of concomitant medications should be given as these may influence some of the analytical measurements or modify metabolic processes.
Case discussion: Round 2 investigations
In our case, VLCFA and white cell enzyme activities were normal. Genetic testing for Alexander disease was negative. Over the next 12 months the patient continued to deteriorate. Discussions were held with the patient's family regarding the possibility of a brain biopsy, principally to exclude the remote chance of an inflammatory disorder, but this was declined. The patient was given a trial of oral steroids, but failed to improve. During this time, mutations in the colony stimulating factor 1 receptor (CSF1R) gene, responsible for HDLS, were identified.45 Sequence analysis of the patient's DNA subsequently identified a single mutation (E847D) in the CSF1R gene, confirming the diagnosis.
The flow diagram in online supplementary figure S2 summarises the investigative process. If no abnormalities are found on biochemical testing or if a diagnosis not known to be associated with any biochemical findings is suspected, then it may be appropriate to proceed to genetic testing. This may be by a single gene analysis or analysis of a panel of candidate genes, depending on clinical suspicion and/or resources. Some centres may also have access to next generation sequencing technologies such as whole exome sequencing, and it is likely that gene chips which can be designed to detect the genetic defects causing a range of different disorders in parallel, in a cost effective manner, will have a major impact on the diagnosis of these and related conditions.46 At time of writing, mutation analysis of the following genes associated with leukodystrophies is available clinically (NHS) within the UK: ABCD1, ARSA, CSA, ERCC6, GALC, GBE1, GFAP, NOTCH3 and PLP1. Further details are available online (http://ukgtn.nhs.uk/). Diagnosis of mitochondrial disorders is coordinated via three specialist centres within the UK (http://www.mitochondrialncg.nhs.uk/).
When should a brain biopsy be considered?
A review of the seven most recent series in evaluating cryptogenic neurological diseases with brain biopsy estimated a yield rate between 29% and 65%.47 The rate was increased to 78% in the Gilkes et al47 series, by biopsing a radiologically identified lesion, in particular with contrast enhancement. Biopsy not only helps in reaching a diagnosis, but can alter clinical management. Rice et al48 found that management was altered as a result of biopsy in 63% of their cases. The modern risk of stereotactic brain biopsy has been estimated at 10–20%,49 with complications including seizures, intracranial infection and haemorrhage, but no long term neurological sequelae. Ideally a panel comprising a neurologist, neurosurgeon and neuroradiologist should discuss the optimal site for biopsy and ensure that the neuropathologist is aware of the clinical phenotype, imaging findings and family history to guide pathological evaluation. Algorithms have been suggested in the literature to aid decision making in presentations such as dementia49 and cryptogenic neurological conditions,47 ,48 but there are none specific to leukodystrophies. We feel that a biopsy in a suspected leukodystrophy should be considered under the following circumstances: the patient is deteriorating and a potentially treatable condition is being considered and/or all other investigations have failed to reach a diagnosis and it could aid in the management of other family members with or at risk of the condition.
Frequently diagnosed adult onset leukodystrophies
We now summarise the clinical presentation and MRI findings for the most frequently diagnosed adult onset leukodystrophies and important non-classical leukodystrophies including CADASIL and mitochondrial conditions. A more detailed discussion of the complexities of mitochondrial disease is beyond the scope of this article, but a number of references are given. When considering a specific leukodystrophy, it should be noted that often the presentation can be non-specific, but certain symptoms and signs and MRI patterns can be used together to help formulate a differential diagnosis and aid investigation.
X-linked adrenoleukodystrophy (X-ALD)
Clinical presentation: Adult onset cerebral ALD usually presents with psychiatric features followed by dementia, ataxia, seizures and death.2 This may occur on the background of adrenomyeloneuropathy (AMN), a slowly progressive spastic paraparesis, neurogenic bladder and bowel dysfunction, sexual dysfunction and peripheral neuropathy.50 Adrenal insufficiency is frequently associated. About 20% of female X linked ALD carriers develop symptoms, usually in their fourth decade, of an AMN-like phenotype, but only very rarely do they have adrenal insufficiency.
MRI features: White matter abnormalities are usually first seen in occipital regions, with early involvement of the splenium of the corpus callosum and posterior limbs of the internal capsule.50 The changes then progress to involve more anterior regions. In cerebral ALD, contrast enhancement at the periphery of the signal abnormalities is said to be characteristic.8 In AMN, the posterior limbs of the internal capsules are frequently involved along with cerebellar and brainstem white matter.51 Spinal cord atrophy may develop. Brain involvement on MRI is very rarely described in women with an ABCD1 mutation. A review of brain MRI imaging in 76 affected women found changes consistent with male patients with cerebral ALD in only two women.52 Although the authors attempted to exclude other known causes of leukodystrophy at the time, the possibility remains that these changes may in fact have been related to a second undiagnosed condition.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
Clinical presentation: Typically, patients present with migraine with a mean age at onset of 30 years;6 ischaemic events usually have their onset in the late 40s. As microvascular changes progress, patients develop cognitive and psychiatric symptoms and seizures.
MRI features: MRI changes usually precede the onset of other symptoms by 10–15 years. The first changes are nodular white matter lesions in the periventricular areas and in the centrum semiovale. The abnormalities then typically become diffuse, symmetrical, and involve the external capsule and characteristically extend into the anterior temporal lobes to involve the temporal pole—a characteristic but not sensitive feature.6
Krabbe disease
Clinical presentation: The adult form (10% of cases) typically presents with pyramidal tract dysfunction and spastic paraparesis.53 ,54 Patients can also develop cognitive decline, seizures and cortical blindness.55 Approximately 20% of patients also have abnormal peripheral nerve electrophysiology, which shows slowing of conduction velocity.56
MRI features: Posterior predominant white matter changes, with sparing of the U fibres and involvement of the splenium of the corpus callosum, are typically seen. A pattern of radiating stripes of normal signal intensity is seen in the cerebral white matter.57 T2 hyperintense changes are seen along the corticospinal tracts58 and the posterior limb of the internal capsule and the pyramidal tracts of the brainstem. On CT, hyperintensities are seen in the thalamus and basal ganglia; it is unclear what these hyperintensities on CT represent, but it has been suggested they may represent minute calcifications.
Cerebrotendinous xanthomatosis (CTX)
Clinical presentation: Typically, patients will present in adolescence with cataracts, but then in adulthood will go onto develop a spastic paraparesis, pyramidal tract signs, cerebellar ataxia, bulbar symptoms and peripheral neuropathy. There may be a childhood history of diarrhoea or failure to thrive. If left untreated, patients can develop progressive dementia and psychiatric symptoms.59 Patients should be examined for other organ involvement including tendon xanthomata (usually involving the Achilles tendon), osteoporosis,60 and respiratory, endocrine and liver involvement.61
MRI features: Non-specific supratentorial atrophy and deep periventricular white matter change can be seen. The U fibres and corpus callosum are spared. The classic picture is high signal intensity within the cerebellar white matter and low signal intensity in the dentate nucleus on T2-weighted imaging.57
Metachromatic leukodystrophy (MLD)
Clinical presentation: The adult form accounts for approximately 20% of MLD presentations.2 The initial symptoms are often psychiatric, followed later by the onset of motor symptoms including spastic paraparesis and cerebellar ataxia,62 and then intellectual and cognitive decline.63 Other neurological signs include optic atrophy and dystonia, a demyelinating peripheral neuropathy62 and late in the disease, seizures.62
MRI features: These include widespread symmetrical confluent white matter change with a periventricular, frontal predominance. Typically there is a (tigroid) pattern of radiating stripes in the affected white matter. The subcortical U fibres are typically spared. Typically, the corpus callosum is involved early.57
Mitochondrial disorders
White matter involvement is increasingly recognised as a common finding in mitochondrial disorders, both those associated with alterations in mitochondrial DNA, which include Leigh disease, Kearns Sayer syndrome, mitochondrial encephalopathy lactic acidosis and stroke-like episodes, mitochondrial neurogastrointestinal encephalomyopathy and Leber's hereditary optic neuropathy, as well as those associated with mutations in nuclear DNA (typically in genes of DNA repair proteins), which are transmitted in a Mendelian pattern. While not traditionally classified as a leukodystrophy, these disorders form an important aspect of the differential diagnosis in this context.
Clinical presentation: Clinical presentation can occur at any stage of life, and many of these disorders have characteristic features, although most present with multi-system involvement.64 Although mitochondrial DNA is inherited by a maternal pattern, no family history may be present, new mutations may arise or heteroplasmy (the variation of mitochondria in the same organism) may result in a higher proportion of mutant mitochondrial DNA in one individual compared with the mother (or siblings inheriting the same mutation), such that the disease manifests earlier or with a different phenotype. Interpretation of the inheritance pattern is made even more difficult by disorders that result from mutations in nuclear DNA.
MRI features: In each of the specific mitochondrial disorders, specific white matter patterns have been described. Lerman-Sagie et al65 provide a useful review of the different white matter patterns in specific disorders. MRI findings suggestive of a mitochondrial disorder include small cyst-like lesions within the abnormal white matter, involvement of both cerebral and cerebellar white matter, and the combination of both white matter change and bilateral basal ganglia lesions.65
Genetic testing: The diagnosis of a mitochondrial disorder involves clinical suspicion, laboratory testing and the use of these results to direct genetic testing. In the first instance, the clinical phenotype should be defined using history, examination, neuroimaging, neurophysiology, audiometry and cardiology and ophthalmological opinion and investigation. Blood tests measuring lactate and cerebrospinal fluid (CSF) examination should also be performed. The next crucial step is a muscle biopsy for which histochemical studies and biochemical analysis of mitochondrial respiratory chain complexes can be performed. These results can then direct the genetic sequencing analyses performed. McFarland et al64 provide a comprehensive overview and algorithm for genetic testing in mitochondrial disorders.
Hereditary diffuse leukoencephalopathy with neuroaxonal spheroids (HDLS)
Clinical presentation: Patients typically present in adulthood, with personality change, and cognitive impairment, particularly executive dysfunction and memory problems.66 Patients then go on to develop spasticity, gait problems (these may be an early feature), ataxia and urinary incontinence.
MRI features: Typically, patients have non-enhancing, symmetrical white matter T2 hyperintensity with a frontal predominance also involving the precentral and postcentral gyrus, extending from the periventricular and deep regions to subcortical tissues. There may be associated atrophy in the regions of hyperintensity and abnormal signal in the corpus callosum with or without atrophy.67 The signal abnormalities extend downwards through the posterior limb of the internal capsule into the pyramidal tracts of the brainstem.57
Adult polyglucosan body disease (APBD)
Clinical presentation: Patients present with a mixed picture of spastic paraparesis, but also have signs of lower motor neurone involvement, and bladder dysfunction. They can also have cognitive impairment, ataxia and extra-pyramidal signs.
MRI features: MRI typically shows diffuse periventricular white matter change involving predominantly the occipital and temporal lobes and the mesencephalon and cerebellum. The white matter changes are most prominent in the periventricular region, posterior limb of the internal capsule and external capsule and spare the U fibres and corpus callosum. In the later stages, thinning of the corpus callosum can develop. There is diffuse cerebral, cerebellar and spinal cord atrophy.68 ,69
Alexander disease
Clinical presentation: In a review of the adult cases reported in the literature (36 cases),17 the most common presentation was with bulbar dysfunction, pyramidal involvement, cerebellar ataxia and sleep abnormalities. Palatal myoclonus, urinary abnormalities and autonomic dysfunction have also been reported.
MRI features: In adult onset disease, there is typically white matter change and atrophy within the medulla oblongata and upper cervical spinal cord. There can be mild to moderate periventricular white matter change, but this is not present in all cases. Other signs suggestive include hyperintensities on T2-weighted images and postcontrast enhancement particularly in patients aged less than 40 years.70 The MRI findings in adult onset Alexander disease differ from those in the infantile/juvenile form which include: extensive white matter change with frontal predominance, a periventricular rim with high signal on T1-weighted images and low signal on T2-weighted images, abnormalities of the basal ganglia and thalami, brainstem abnormalities and contrast enhancement.71
Adult onset autosomal dominant leukodystrophy (ADLD)
Clinical presentation: Typically, patients present in the fourth or fifth decade with autonomic abnormalities, followed by pyramidal symptoms, ataxia and cognitive deterioration.19
MRI features: Diffuse white matter T2 hyperintensities involving the frontal lobe, parietal lobe and middle cerebellar peduncle. There is atrophy of the brainstem and corpus callosum.
Vanishing white matter disease (VWM)
Clinical presentation: At least 25 adult onset cases have been described in the literature.20 Features include spasticity, cerebellar signs, seizures and dementia. A third of patients presented with psychiatric symptoms.20 Female patients can also present with primary or secondary ovarian failure.
MRI features: MRI changes are characterised by diffuse T2 hyperintensity and hypointensity with associated cystic change (seen on FLAIR imaging) and atrophy. There is also corpus callosal atrophy and T2 and flair hyperintensities. Atrophy of the cerebellar hemispheres has also been reported, both with and without associated white matter changes.20 In end stage, vanishing white matter disease cerebral hemispheric white matter may have vanished leaving a ventricular wall and cortex, with little in between.57
Pelizaeus-Merzbacher disease (PMD) and Pelizaeus-Merzbacher-like disease
There are limited case reports in adults; however, Pelizaeus–Merzbacher disease (PMD) is often on the differential diagnosis list due to the common presentation of spastic paraplegia.
Clinical features: The adult type may present with a chronic neurological syndrome including tremor, ataxia and dementia.32 Heterozygote females are usually asymptomatic, but can present with gait abnormalities. There are reports of heterozygote females in their 40s developing clumsiness, excessive falling and gait disturbances (wide based gait), hyper-reflexia, tremor and extensor plantar responses.72 Hereditary spastic paraplegia type 2 is clinically distinct from PMD, but is also due to mutations in the proteolipid protein (PLP) gene. Pelizaeus–Merzbacher-like disease is clinically indistinct from PMD; however, patients are negative for the PLP mutation.
MRI features: Varying MRI patterns have been reported including diffuse white matter change, both with and without involvement of the corticospinal tracts, and patchy changes involving the cerebral hemispheres.73
This review highlights that there are a wide range of leukodystrophies.57 New genetic findings are being identified with advanced techniques, but many are as yet undiagnosable in life. OMIM74 is a useful resource for newly characterised leukodystrophies.
Are there any specific treatments for adult onset leukodystrophies?
Support is required for patients and families during the process of making a diagnosis and once it is made. Due to the late onset, many affected patients have already had their own children, and detailed genetic counselling is required to explain the full implications of a particular diagnosis.
The treatment of the leukodystrophies in adulthood is largely supportive. Even if specific treatment is available, by the time a diagnosis is made the condition in many adults may be very advanced. For example, chenodeoxycholic acid has been used as therapy for cerebrotendinous xanthomatosis since 1975. However, after significant neurological disease is established, the effect of treatment is limited and deterioration may continue.75 ,76
Nonetheless, this is a very active area of research and a number of clinical trials are currently underway worldwide. These include haematopoietic stem cell transplant (ALD, MLD, Krabbe disease, Tay–Sachs disease), human placental-derived stem cell transplant (ALD, MLD, Krabbe disease), intrathecal enzyme replacement therapy (MLD), intracerebral gene therapy (MLD), haematopoietic stem cell gene therapy (MLD), allogeneic bone marrow transplant (ALD, MLD, Krabbe disease) and autologous haematopoietic stem cell transplant transduced with ABCD1 lentiviral vector (ALD). Further details can be found on http://www.clinicaltrials.gov, but it must be noted that not all studies are recruiting adult patients.
Conclusions
Adult onset leukodystrophies represent a highly complex area of adult neurology, compounded by their rarity, with a total prevalence of 300 cases/million. There is a wealth of information on paediatric leukodystrophies, but adult onset cases can be misdiagnosed as atypical MS or young onset vascular or neurodegenerative dementia.66 In this overview, our aim is to describe a logical approach to the diagnostic process, reducing unnecessary (and expensive) investigations and using selectively the newer genetic advances.
Data sharing statement
The authors would be happy to be contacted to discuss or see patients with possible adult onset leukodystrophies.
Acknowledgments
The authors would like to acknowledge the UK National Institute of Health Research (NIHR) University College London Hospitals Biomedical Research Centres funding scheme, the NIHR Queen Square Dementia Biomedical Research Unit, The Medical research council (MRC) and Wellcome Trust. The Dementia Research Centre is supported by Alzheimer’s Research UK, the Brain Research Trust and the Wolfson Foundation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure
Footnotes
-
Contributors RA, NF, JC: manuscript concept, manuscript writing and preparation; EM: manuscript writing and preparation; ID: manuscript writing and preparation, figure preparation and radiological input; MP, JS, CM, RL, HH, JR: manuscript writing and preparation.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.