Article Text

Abstract
Objectives Alemtuzumab is a newly licensed treatment of active relapsing-remitting multiple sclerosis (RRMS) in Europe, which in phase II and III studies demonstrated superior efficacy over β-interferon in reducing disability progression over 2–3 years. In this observational cohort study, we sought to describe our longer-term experience of the efficacy and safety of alemtuzumab in active RRMS.
Methods Clinical and laboratory data including serial Expanded Disability Status Scale (EDSS) assessments, from all 87 patients treated with alemtuzumab on investigator-led studies in Cambridge, UK, from 1999 to 2012, were collected. The occurrence of adverse events including secondary autoimmunity, malignancy and death, and pregnancy outcomes was recorded. Baseline variables including age, disease duration and relapse rate were compared in univariate and logistic regression analyses between groups with different disability outcomes.
Results Over a median 7-year follow-up (range 33–144 months), most patients (52%) required just two cycles of alemtuzumab. In the remaining patients, relapses triggered re-treatment to a total of three cycles (36%), four cycles (8%) or five cycles (1%). Using a 6-month sustained accumulation of disability definition, 59/87 (67.8%) of patients had an improved or unchanged disability compared with baseline. By an area under the curve analysis, 52/87 (59.8%) patients had an overall improvement or stabilisation of disability. Higher baseline relapse rate was associated with worse long-term disability outcomes, with trends for longer disease duration and older age at first treatment. Secondary autoimmunity was the most frequent adverse event occurring in 41/86 (47.7%) patients, most commonly involving the thyroid gland.
Conclusions Alemtuzumab is associated with disease stabilisation in the majority of patients with highly active RRMS over an average seven-year follow-up. No new safety concerns arose over this extended follow-up.
- Multiple Sclerosis
- Immunology
Statistics from Altmetric.com
Introduction
In September 2013, alemtuzumab was licensed in the European Union as a treatment for active multiple sclerosis. As it is taken up into general neurological practice, there is a need for information on its long-term dosing, safety and efficacy.
Alemtuzumab, a humanised monoclonal antibody that targets the CD52 antigen expressed on the surface of lymphocytes, monocytes and natural killer cells (NK cells), is given as cycles of treatment, over 5 days initially and over 3 days for subsequent pulses. Each infusion causes pan-lymphocyte depletion, with variations in the rate and extent of lymphocyte subset reconstitution leading to modifications of the lymphocyte repertoire lasting several years.1 ,2
In its pivotal phase II and III trials, alemtuzumab significantly reduced the risk of relapse and accumulation of disability compared with interferon β-1a over 2–3 years.3–5 Its principal adverse effect is secondary autoimmunity affecting the thyroid gland in 30% of patients but less commonly causing immune thrombocytopenic purpura (ITP) (1–3%).3–5 ,6 Limited data emerged from two further years of follow-up of a subgroup (198/334) of patients in the phase II trial in which superior efficacy over β-interferon was maintained with only eight patients requiring a further cycle of treatment.7
In this observational cohort study, we summarise our total experience from 1999 to 2012 of treating relapsing-remitting multiple sclerosis with alemtuzumab in two rater-blinded investigator-led open-label studies at a single site with 100% follow-up. We present disability and safety data recorded over a longer period (average 7 years) than any previous study.
Methods
Patients and assessments
All (n=87) patients with relapsing-remitting multiple sclerosis were followed at our site (under an approved protocol; Research Ethics Committee number 11/ee/0007) from two single-arm, open-label, rater-blinded, investigator-led studies, whose early results have been reported: 67 on the ‘CAMMS224’ trial (1999–2010; Research Ethics Committee number 03/07) and 20 on the ‘SM3 trial’ (2005–2008; REC 05/Q0501/64).8 ,9 Inclusion criteria for both were relapsing-remitting multiple sclerosis; ≥1 relapse in the preceding year; Expanded Disability Status Scale (EDSS) <6.0; disease duration <10 years; without previous exposure to experimental therapy. Data were censured on 31 May 2012. Patients were seen quarterly for 2 years following each cycle of alemtuzumab, 6-monthly over the next 2 years, at least annually thereafter and within a week of reporting new symptoms. Disability was assessed using the EDSS by a rater blinded to treatment and previous EDSS scores.10 Laboratory investigations performed at each visit included a full blood count (FBC), lymphocyte subsets, thyroid function tests, anti-nuclear antibody (ANA) and antithyroid peroxidase autoantibodies.
Treatments
Two cycles of alemtuzumab were administered 12 months apart; further cycles were offered if a relapse occurred. Alemtuzumab was given by intravenous infusion on five consecutive days at baseline and on three consecutive days for subsequent cycles. The initial dose (20 mg/day) was increased to 24 mg/day in 2003 following a change in supplier. From 2006, the dose was reduced to 12 mg/day to align with the phase III study dosing. Infusion-associated symptoms were reduced by corticosteroid pretreatment; most patients also required antihistamines and paracetamol.11 Patients in the SM3 trial also received a biologically inert variant of alemtuzumab, which prevented antialemtuzumab antibodies.9 Patients received no other disease-modifying therapy.
Disability outcomes
Sustained accumulation of disability (SAD) was defined as an increase in EDSS, sustained for at least 6 months of ≥1.5 EDSS points if the baseline EDSS was 0; ≥1.0 point if the baseline EDSS was ≥1 but <5.5; and >0.5 points if the baseline EDSS was ≥5.5. Sustained reduction in disability (SRD) was defined as a reduction in the EDSS score of either ≥1.0 or 0.5, for baseline EDSS scores below and above 5.5 respectively sustained for at least 6 months. In keeping with its previously published definition, analysis for a SRD was restricted to those with a baseline EDSS ≥2.0.12 SAD and SRD sustained for 6 and 12 months were used to define events for a Kaplan–Meier (KM) analysis.
The area under the EDSS/follow-up time curve (AUC) was calculated as per the trapezium method correcting for baseline disability and rescaling—with changes of 0.5 for EDSS scores ≥5.5 and <7.0 being normalised to a 1.0 point change at all other levels of the scale.13–15 Individual patients were categorised as (i) ‘net improved’ for an AUC of <−0.5 EDSS-years; (ii) ‘net worse’ for an AUC > +0.5 EDSS-years and (iii) ‘net unchanged’ for an AUC between −0.5 and 0.5 EDSS-years. Each patient was also categorised, by two independent assessors, into one of six descriptive disease course categories as defined by Liu et al from the profile of their plot of EDSS-change versus follow-up time. Under this classification system, SRD and SAD became ‘sustained improvement’ and ‘sustained progression’ if the disability change was maintained until last follow-up or, if not, as ‘erroneous improvement’ and ‘erroneous progression’.16 ‘Minimal change’ indicated an EDSS change ≤0.5 points from baseline at all measurements made over the course of follow-up. The remaining profiles that did not fit any of the above categories were labelled as ‘fluctuating’. From these six categories, three groups were defined: ‘confirmed stable’ (‘sustained improvement’ or ‘minimal change’); ‘unsustained change’ (‘erroneous progression’, ‘erroneous improvement’ or a ‘fluctuating’ course); and ‘confirmed worsening’ comprising those with ‘sustained progression’. We defined secondary progression as two consecutive SAD events, the second from the new EDSS baseline established after the first SAD event, and in which the increase in EDSS occurred independent of relapses. From 2002 until March 2011, the same blinded Neurostatus-certified rater performed every disability assessment.17
Safety follow-up
Information on infections and symptoms of relevant organ-specific autoimmunity was enquired about at each outpatient clinic review. In addition, patients were encouraged to contact the trial team to report any new symptoms and were counselled about urgent reporting of the signs and symptoms of ITP. From September 2005, in addition to the FBC testing performed at each clinic visit, monthly platelet counts were performed for 3 years after each alemtuzumab cycle. Secondary autoimmunity was defined as new symptomatic autoimmune disease; sustained abnormal thyroid stimulating hormone, free thyroxine or tri-iodothyronine levels; or novel serum autoantibodies on ≥2 occasions ≥3 months apart.
Statistical analyses
Between groups with and without SAD: unpaired t tests were used to compare baseline variables of age and disease duration; differences in baseline EDSS were tested by the Mann–Whitney U test and prior disease-modifying therapy use by the Pearson's χ2 test. These variables were also analysed in a logistic regression model with SAD as dependent variable. The Poisson test was used to compare pretreatment and post-treatment relapse rates. Univariate analyses were used to compare the same baseline variables between patients with ‘net worse’ versus combined ‘net better’ and ‘net unchanged’ AUC scores. To compare the SAD/SRD and AUC techniques of disability classification, a 3×3 contingency table was constructed for ‘worse’, ‘unchanged’ and ‘improved’ disability and analysed.18
Data were analysed using the Predictive Analytics Software package (PASW Statistics 18, SPSS Inc, Chicago, USA).
Results
Patients and treatments
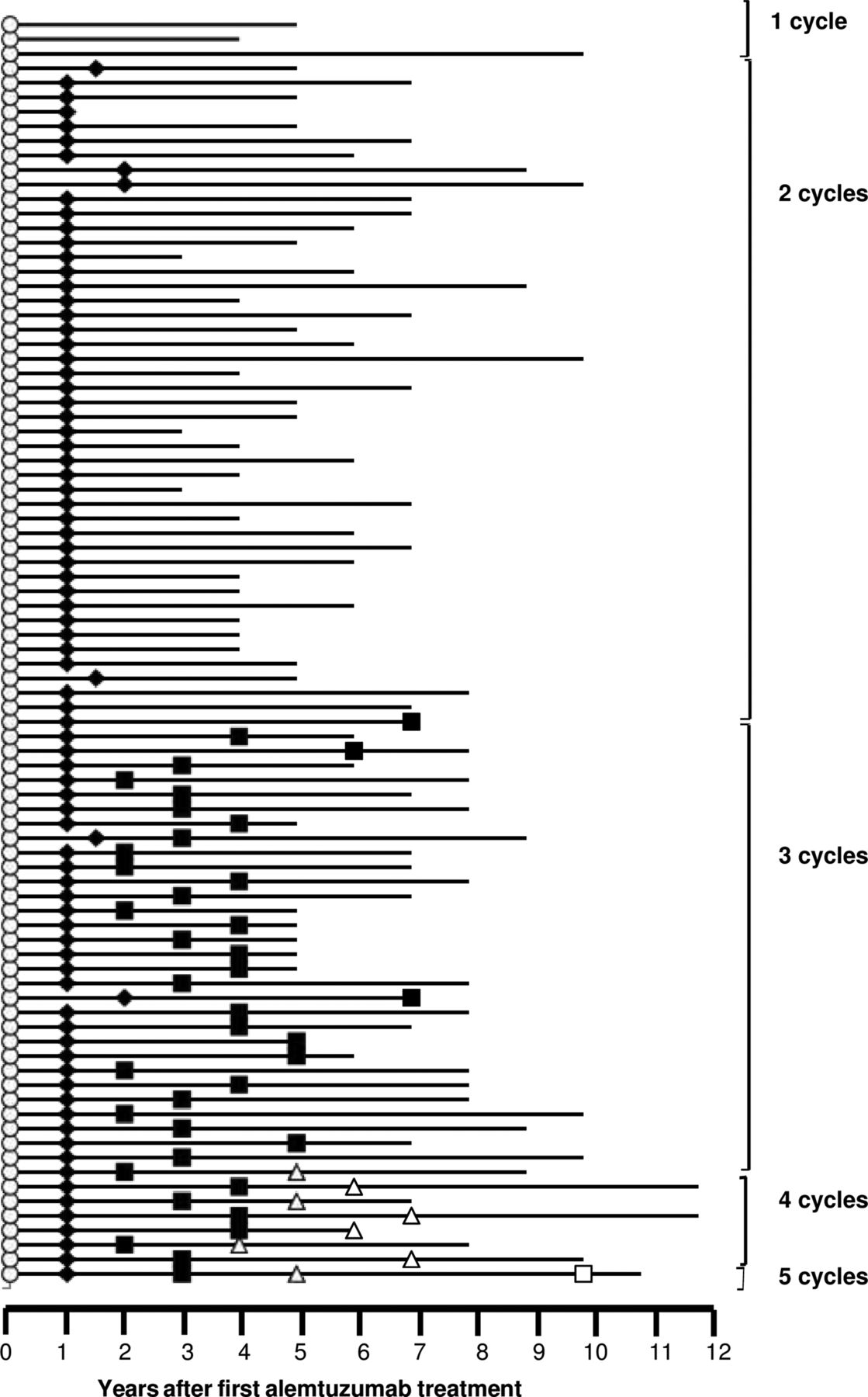
Baseline characteristics are given in table 1. Total follow-up to last-recorded EDSS was 559 patient-years, and 624 patient-years to last clinic visit. Most patients (45; 52%) received just the two planned cycles of alemtuzumab, 12 months apart. Further cycles were offered if a relapse occurred: 31 patients (36%) received three; 7 patients (8%) four; and 1 patient five cycles (figure 1). Three patients (4%) received only one treatment cycle; one developed Goodpasture's disease and was advised against re-treatment; and two patients declined further cycles. Median intervals between treatments were 26 months (n=39, IQR 17.5–35 months) for cycles 2–3; 26 months (n=8, IQR 23–33 months) between cycles 3 and 4; and 54 months (n=1) for cycles 4–5.
Baseline characteristics and follow-up of 87 patients with relapsing-remitting multiple sclerosis treated with alemtuzumab

Follow-up and retreatment of 87 patients after alemtuzumab.
Clinical efficacy outcomes
Mean annualised relapse rate after alemtuzumab, recorded prospectively over all follow-up, was 0.16 (SD 0.26) compared with 1.78 (SD 0.82), assessed retrospectively, for the two pretreatment years. Mean EDSS at baseline was 3.8 (SD 1.94) and at last follow-up was 3.6 (SD 2.30) (p=0.56, t test). A greater proportion of eligible patients had a 6-month SRD than a 6-month SAD; 30/69 (43.5%) versus 28/87 (32.2%) (omitting patients with baseline EDSS <2.0 from the SRD analysis; table 2 and figure 2). Similarly, when the minimum time interval used to define SAD or SRD events was extended to 12 months, more patients recorded a 12-month SRD event than a 12-month SAD event; 26/69 (37.7%) versus 19/87 (21.8%).
Baseline EDSS, length of follow-up and disability outcomes based on the area under the curve of the EDSS versus follow-up time plot of n=87 alemtuzumab-treated patients.

Kaplan–Meier plots of sustained accumulation of disability and sustained reduction in disability events sustained at 6 months (A, B) and 12 months (C, D) in alemtuzumab-treated patients.
Applying the AUC analysis method, median disability improved following alemtuzumab by −0.46 EDSS-years (IQR −6.97–2.90), with 43 patients having a ‘net improved’ disability (AUC value <−0.5), 35 patients having a ‘net worse’ disability (AUC value >0.5) and 9 patients having a ‘net unchanged’ disability (AUC value ≥−0.5, ≤0.5) by the end of follow-up (table 2 and figure 3). Under the AUC-based category and group analyses, 20 patients had ‘sustained progression’ while 17 had a ‘sustained improvement’ (figure 4). In seven patients, the increase in EDSS score associated with a 6-month SAD event was not sustained throughout follow-up, that is, ‘erroneous progression’, while the disability improvement recorded as a 6-month SRD event was not sustained over all follow-up in 15 patients, that is, ‘erroneous improvement’. The EDSS scores of seven patients remained within 0.5 EDSS points of their baseline score in the ‘minimal change’ category, while the remaining patients had a disability change profile that was classified as ‘fluctuating’. Collapsing these categories into three disability-change groups resulted in 24/87 patients (27.6%) having ‘confirmed stable’ disability; 20/87 (23%) having ‘confirmed worsening’ and 43/87 (49.4%) having an ‘unsustained change’. Four alemtuzumab-treated patients (5%) fulfilled the definition of secondary progression of two consecutive SAD events.

Plot of individual patient area under the curve values calculated from the plots of Expanded Disability Status Scale change from baseline versus follow-up time.

Disease course categories of 87 alemtuzumab-treated patients as per Liu et al.
There was overall moderate agreement between the SAD and AUC methods in categorising patients as net worse, net better or unchanged (κ 0.55; 95% CI 0.42 to 0.68; table 3). Proportions of agreement in the net worse or net better category were 70% (95% CI 53% to 84%) and 62% (95% CI 47% to 76%), but much lower in the unchanged category at 23% (95% CI 0.10% to 42%) because patients with a ‘fluctuating’ disease course tended to be categorised net worse or net better by AUC and unchanged by the SAD method.
A comparison of results from two disability outcome measures applied to alemtuzumab-treated patients
To determine whether baseline characteristics predicted disability outcome after alemtuzumab treatment, we performed univariate analysis of baseline variables in patients with unfavourable and favourable disability outcomes, and binary logistic regression with disability worsening as dependent variable, using SAD/SRD and AUC data (see online supplementary tables S1 and S2). Across these approaches, poor disability outcome was associated with high pretreatment relapse rate. The mean number of relapses in the 2 years before treatment was 4.11 (SD 1.73) in the SAD group versus 3.23 (SD 1.68) in the SRD group (p<0.001 Poisson test). There were trends in univariate analyses suggesting poor outcome was associated with older age and, less convincingly, longer disease duration at time of treatment (see online supplementary table S1).
Secondary autoimmunity
Clinical autoimmune disease developed in 41 patients (48%, omitting one patient with pre-existing thyroid disease); a further 12 (14%) patients developed sustained novel autoantibodies (9 ANA and 3 anti-TPO) with no evidence of associated clinical disease. This occurred a median of 32 months after first treatment (IQR 19.5–42.0) and a median 16 months since last treatment (IQR 9.5–23.0 months) (figure 5). Autoimmunity was not associated with the number of alemtuzumab treatment cycles administered (p=0.457, Mann–Whitney U test).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Occurrence of clinical autoimmunity after alemtuzumab timed from first (A) and most recent (B) treatment.
Thyroid autoimmunity developed in 35/86 (41%) of whom 22/35 (63%) had hyperthyroidism (Graves’ disease); 1 patient had transient thyroiditis, and 12 (34%) developed primary hypothyroidism with positive antithyroid peroxidase antibodies. Most patients were treated medically; three with Graves’ disease also required radio-iodine treatment.
Three patients (3.5%) developed ITP: case 1 (a 19-year-old man at first treatment) noticed easy bruising 43 months after his third alemtuzumab cycle. An FBC revealed a platelet count of 72×109/L (reference range 150–400×109/L); eight days later this had recovered to 123×109/L without treatment. Subsequent platelet counts have remained stable. This patient also developed Graves’ disease 12 months after first alemtuzumab treatment. Case 2 (a 27-year-old man at first treatment) was noted to have mildly reduced platelet counts on routine blood testing (ranging from 86 to 120×109/L), 9 months after his third cycle of alemtuzumab. A year later his platelet count fell to 12×109/L and he was treated with IV immunoglobulin (IVIg) and pulse steroids, followed by rituximab. He failed to tolerate rituximab due to an infusion reaction. Six years after it was first noted, and on no regular medication, his thrombocytopenia persists, with pulse steroids administered for platelet counts <20×109/L. Case 3 (a 25-year-old female at first treatment) developed ITP 1 month after her second alemtuzumab treatment. She received IVIg and steroids when her count fell to 2×109/L. Oral steroids were tapered over 4 months and her platelet count has since remained stable (37 months from onset) with platelet counts ranging from 110 to 150×109/L.
Asymptomatic autoimmune neutropenia developed in a single patient 3 months after her second alemtuzumab cycle, with a neutrophil count of 0.5×109/L. Her neutrophil count rose to within normal range over 6 months without treatment. Antigranulocyte antibodies, which were not present in a pretreatment serum sample, were detected during the period of neutropenia. One patient developed autoimmune haemolytic anaemia, which was detected on FBC monitoring and whose case has been previously reported.9 A case of Goodpasture's disease requiring renal transplantation has also been reported previously in a separate publication. This patient remains well on long-term immunotherapy postrenal transplant.8
Infections
Eleven cases of varicella zoster virus reactivation, including two herpes zoster ophthalmicus and one Ramsay–Hunt syndrome, occurred all involving a single dermatome. There was one case of primary varicella zoster virus infection. The mean interval from most recent treatment to varicella infection was 12 months (SD 13.5 months). There were no serious infections that required hospitalisation. It is likely that minor infections were not reported by patients at longer follow-up intervals.
Pregnancies
A total of 15 babies were born to 12 women treated with alemtuzumab during the course of the follow-up period after a median interval from most recent treatment of 26 months (range 13–86 months); they had received a median of two cycles of alemtuzumab and a mean cumulative dose of 174 mg (SD 52 mg). All deliveries and births were uncomplicated. Six males fathered seven live births, a median of 14 months (range 8–44 months) from most recent treatment to conception. The partner of another male patient had a miscarriage at 17 weeks gestation, while a subsequent live birth to the same couple was diagnosed with a congenital heart defect requiring surgical repair. No patient developed subfertility after alemtuzumab.
Malignancies
No malignancies were noted. One case of Castleman's disease, a pre-lymphomatous condition, is reported elsewhere.9
Lymphopenia
As previously described, monocyte numbers recovered rapidly, whereas T cells reconstituted slowly after alemtuzumab.1 ,6 At last visit, mean CD4 and CD8 counts were in the normal range (see online supplementary figure S1). A detailed account of the dynamics of lymphocyte reconstitution is reported elsewhere.19
Discussion
This is the longest experience of alemtuzumab treatment of multiple sclerosis. We conclude that, in patients with early active relapsing-remitting disease (median EDSS 3.5, median disease duration 3 years), two cycles of alemtuzumab, with up to three further cycles triggered by a relapse, lead to a stabilisation of disability in the majority of patients treated over an average 7-year follow-up. Over a third of the cohort were considered to have failed previous disease-modifying therapy, mainly β-interferon or glatiramer acetate with no difference in outcomes observed between this group and untreated patients, suggesting alemtuzumab may be effective as both first-line and second-line therapy of active multiple sclerosis, which is the indication approved by the European Union. A consistent finding between analyses employing different disability outcome measures was that higher baseline relapse rate was associated with a worse long-term disability outcome. A trend for older age and longer disease duration at treatment to associate with an adverse disability outcome lends support to the early use of alemtuzumab during a hypothetical ‘window of therapeutic opportunity’ in patients with active disease.8
Using the conventional 6-month SRD or SAD definition of disability change, 59/87 (67.8%) patients had an improved or unchanged disability when last assessed compared with baseline. However, this approach omits significant disability changes after the SAD/SRD event, which may occur in up to 25% of patients.20 These are captured by the AUC analysis: by this measure, 52/87 (59.8%) patients had an overall improvement or stabilisation of disability. There was reasonable agreement between categorisation of disability change using the AUC or SRD/SAD methods, the main difference being that fewer patients were described as unchanged using SRD/SAD.
The limitations of this open-label, single-centre cohort are the absence of contemporaneous controls and the variability in patient follow-up times. However, no patient was lost to follow-up and inter-rater variability in EDSS score assignment was minimised by having one blinded rater perform all EDSS assessments over 9 years of follow-up. This ‘real-world’ experience provides additional information to the pivotal trials needed to evaluate the complex interplay of efficacy, safety, convenience and timing of exposure for a newly licensed therapy. In addition in this study, we addressed the issue of secondary progression, which is not a routine outcome in treatment trials due to their short duration. We propose an operational definition based on consecutive discrete 6-month SAD events in the absence of relapses. Using this definition, just 4 of our 87 patients (5%) developed secondary progression.
Secondary autoimmunity rates in this study (48%) are higher than previously reported, probably reflecting the longer follow-up. Most cases occurred within 2 years of last alemtuzumab cycle, after which their frequency fell with no cases in this series observed after 5 years from the time of the last alemtuzumab cycle. Most autoimmunity involved the thyroid gland and was responsive to standard medical management. One of the three ITP cases was unusual, compared with experience in other trials, in having a chronic stable asymptomatic course, off treatment. Our recent work has identified pretreatment serum IL-21 levels, and marked homeostatic lymphocyte proliferation, as factors associated with the development of post-alemtuzumab autoimmunity.19 ,21 It is hoped that this work will allow the prediction of alemtuzumab-related secondary autoimmunity in the future, but currently appropriate assays are not available.22 No adverse events were recorded during the pregnancies or deliveries of the 15 babies born to mothers who had been treated with alemtuzumab. No new or late-occurring adverse events were recorded outside of those previously reported in the literature.
Taken together, these data show that over a median of 7 years, most people with early, active relapsing-remitting multiple sclerosis treated with alemtuzumab experience a stabilisation of disability. Secondary autoimmunity remains the most frequently reported adverse event post-treatment.
Acknowledgments
AJC is funded by the NIHR Cambridge Biomedical Research Centre. JLJ is a NIHR clinical lecturer. Clinical trials are performed at the Wellcome Trust Clinical Research Facility. The SM3 trial was funded by the Moulton Charitable Foundation. Priscilla Loh kindly assisted with the statistical analyses.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Correction notice Since this paper was published online an additional author (Michael Fahey) was added to the author list. The acknowledgment statement was also updated to include thanks to Pricilla Loh.
-
Funding AJC and OT are funded by the NIHR Cambridge Biomedical Research Centre. JJ is a NIHR clinical lecturer. Clinical studies were performed at the Wellcome Clinical Research Facility.
-
Contributors OT collected and analysed data and wrote the manuscript. LC performed data analysis, data interpretation and writing of the manuscript. GH-C performed data collection and interpretation. IB performed blinded EDSS assessments. KH and NR reviewed the manuscript. KM, TB, LA and OK-E reviewed the manuscript. JJ and AC performed data analysis and interpretation and writing of the paper. DASC reviewed the paper. All authors reviewed the manuscript.
-
Competing interests NR reports receiving consulting fees from Genzyme (a Sanofi company). JJ reports receiving consulting, and lecture fees from Bayer Schering Pharma and from Genzyme (a Sanofi company). DASC reports receiving consulting fees, lecture fees and grant support from Genzyme (a Sanofi company) and lecture fees from Bayer Schering Pharma, and has consulted for Lundbeck, all on behalf of the University of Cambridge. AC reports receiving consulting and lecture fees from Genzyme (a Sanofi company), lecture fees from Merck Serono and research support paid to his institution from Genzyme (a Sanofi company).
-
Ethics approval Ethical approval was provided by the local Research Ethics Committee: SM3 REC 05/Q0501/64, CAMMS224 REC 03/078, CAMSAFE study REC 11/ee/0007.
-
Provenance and peer review Not commissioned; externally peer reviewed.