Article Text

Abstract
Background The international Inherited Neuropathy Consortium (INC) was created with the goal of obtaining much needed natural history data for patients with Charcot-Marie-Tooth (CMT) disease. We analysed clinical and genetic data from patients in the INC to determine the distribution of CMT subtypes and the clinical impairment associated with them.
Methods We analysed data from 1652 patients evaluated at 13 INC centres. The distribution of CMT subtypes and pathogenic genetic mutations were determined. The disease burden of all the mutations was assessed by the CMT Neuropathy Score (CMTNS) and CMT Examination Score (CMTES).
Results 997 of the 1652 patients (60.4%) received a genetic diagnosis. The most common CMT subtypes were CMT1A/PMP22 duplication, CMT1X/GJB1 mutation, CMT2A/MFN2 mutation, CMT1B/MPZ mutation, and hereditary neuropathy with liability to pressure palsy/PMP22 deletion. These five subtypes of CMT accounted for 89.2% of all genetically confirmed mutations. Mean CMTNS for some but not all subtypes were similar to those previously reported.
Conclusions Our findings confirm that large numbers of patients with a representative variety of CMT subtypes have been enrolled and that the frequency of achieving a molecular diagnosis and distribution of the CMT subtypes reflects those previously reported. Measures of severity are similar, though not identical, to results from smaller series. This study confirms that it is possible to assess patients in a uniform way between international centres, which is critical for the planned natural history study and future clinical trials. These data will provide a representative baseline for longitudinal studies of CMT.
Clinical trial registration ID number NCT01193075.
- GENETICS
- NEUROGENETICS
- NEUROPATHY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Inherited peripheral neuropathies, collectively known as Charcot-Marie-Tooth disease (CMT), are among the most common inherited neurological diseases with a prevalence of 1 in 2500 individuals.1 They encompass a clinically heterogeneous set of disorders, and vary greatly in severity, spanning a spectrum from mildly symptomatic forms to those resulting in severe disability. Patients typically present with length-dependent weakness, atrophy and sensory loss. CMT is also known as hereditary motor and sensory neuropathy (HMSN). Hereditary motor neuropathy (HMN) and hereditary sensory neuropathy (HSN) are related disorders and can also be considered as part of the CMT family. Over 80 causative genes of CMT have been identified and many more remain unknown.2 The natural history of these various forms of CMT remains poorly understood, at least in part, because these are rare disorders and individual centres do not follow enough patients to perform natural history studies. Furthermore, validated clinical instruments for measuring disease severity have become available only recently and have not yet been employed in many of the rare CMT subtypes.
The Inherited Neuropathies Consortium (INC) is a member of the Rare Diseases Clinical Research Network (RDCRN) and was created in part to perform natural history studies in CMT. The INC currently includes 17 sites, (14 in the USA, and one each in the UK, Italy and Australia). The natural history study of the INC records clinical, electrophysiological and genetic data from patients evaluated. In this paper, we report patients evaluated from the initial 13 sites (10 in the USA, and one each in the UK, Italy and Australia) to ascertain the subtype frequencies and the baseline clinical severity of each subtype using validated clinical outcome measures. These quantifiable clinical data add to the literature in providing the clinical severity of a variety of CMT subtypes and also act as a baseline for a longitudinal natural history study of CMT subtypes, a prerequisite for clinical trials.
Materials and methods
Patient data for the INC natural history study of CMT were collected on 1652 patients between April 2009 and 2013 at 13 sites within the INC. All patients were examined by clinical investigators who had received training in the proper use of the CMT Neuropathy Score (CMTNS), a 36 point composite score based on patients’ symptoms (3 items), signs (4 items) and neurophysiology (2 items). On the basis of the CMTNS, patients can be classified as having mild, moderate or severe disease (CMTNS of <10, 11–20 or >20, respectively). Patients in the severe category often require a walker or wheelchair. Patients with moderate scores frequently rely on ankle orthotics but walk independently, and patients in the mild range have little walking difficulty aside from occasional tripping. Participants who did not have neurophysiology studies received a CMT Examination Score (CMTES), which is the CMTNS without neurophysiology. Therefore, the maximum CMTES is 28 rather than 36.3
Patients were considered to have CMT if they had a sensory and/or motor peripheral neuropathy and a family history of a similar condition. Sensory and/or motor neuropathies were diagnosed based on the presence of length-dependent sensory loss (small and large fibre modalities), weakness and atrophy as well as decreased deep tendon reflexes particularly at the Achilles tendon. Nerve conduction studies were also used to confirm neuropathy in most patients. Patients without a family history whose clinical history and electrophysiological findings were consistent with CMT were also included. First-degree and second-degree relatives of patients with a confirmed genetic mutation who demonstrated a similar clinical phenotype and electrophysiological findings were assumed to have the same mutation. Patients with a history of an acquired peripheral neuropathy were excluded.
Data collected included the type of CMT (CMT1, CMT2, HMN, HSN, hereditary neuropathy with liability to pressure palsy (HNPP)), subtype of CMT (CMT1A, CMT1B, etc) and the pathogenic mutation if known. If the genetic diagnosis was not known at the initial evaluation but was determined at a later time point, the diagnosis was recorded and included in the analysis. Patients’ CMTNS or CMTES were recorded at each visit. If patients did not undergo electrophysiology at a particular visit, the CMTES was obtained alone. Standard methods were used for all electrophysiology, and all sensory responses were antidromic. A Clinical Laboratory Improvement Amendments (CLIA) certified laboratory in the USA or an equivalent certified testing facility outside of the USA performed all genetic testing. All sites participating in protocol 6601 received Institutional Review Board (IRB)/Ethics Board approval for the study. All patients or their guardian signed consent forms. This trial was registered at clinicaltrials.gov (ID number NCT01193075).
Results
Distribution of patients
Baseline data were obtained from the initial 1652 patients from protocol 6601. Four hundred and twenty-three participants were 18 years of age or less. In total, 1427 patients were given a clinical diagnosis of CMT. Of those patients in whom a subtype of CMT was specified, 910 were classified as having CMT1, 237 as having CMT2, 37 as having CMT4 (recessive demyelinating or axonal forms of CMT), 107 as having CMT1X, 47 as having HMN, 53 as having HSN and 36 as having HNPP (figure 1). 997 patients (60.4% of total patients enrolled) received a genetic diagnosis. Of the total patients with CMT1, 836 (91.2%) received a genetic diagnosis. In contrast, only 43% of patients with CMT2 received genetic confirmation.
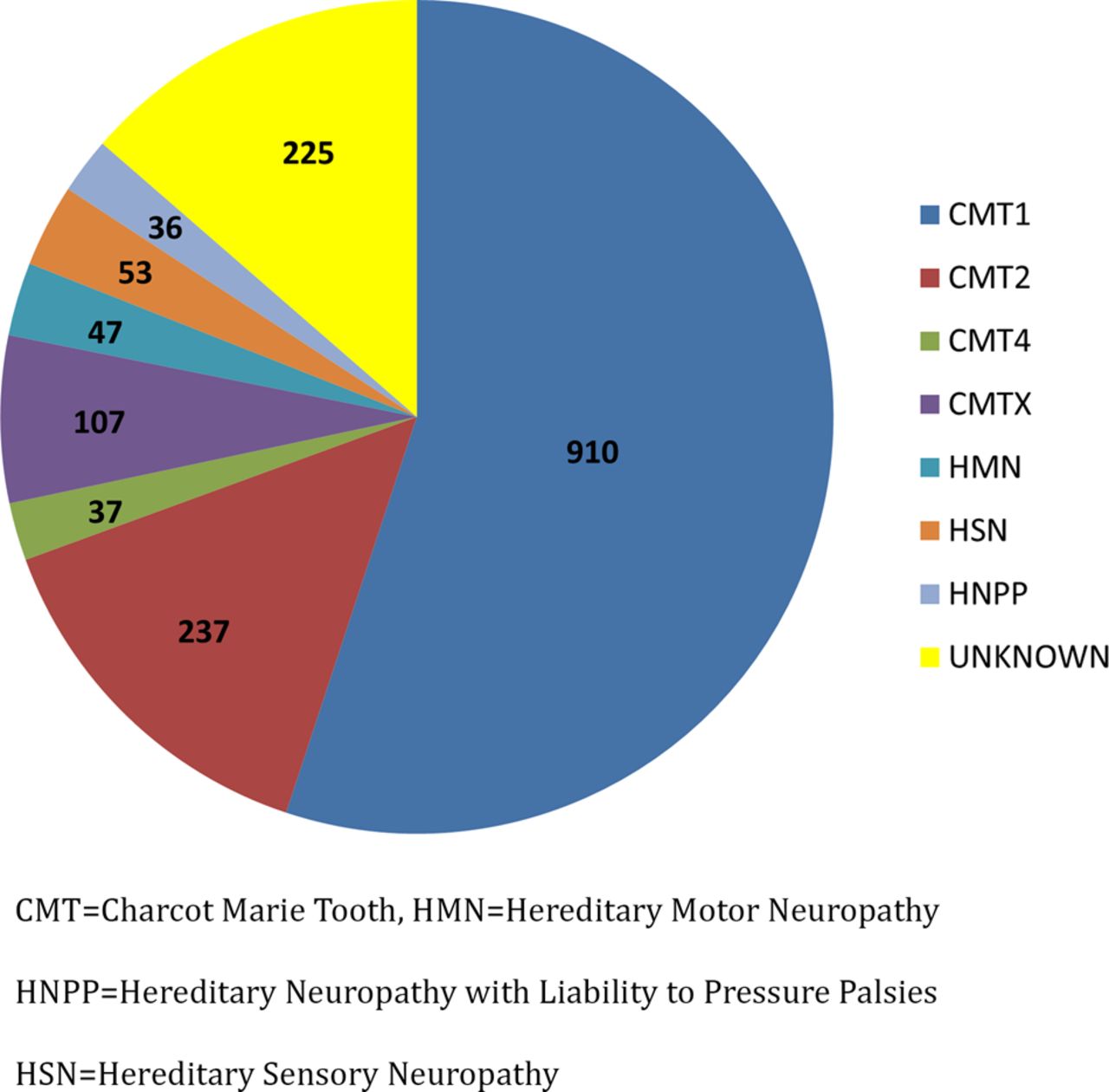
{kind=link}
Distribution of clinically determined Charcot-Marie-Tooth (CMT) subtypes.
The most common genetic diagnoses for our patients were CMT1A (PMP22 duplication), CMT1X (GJB1 mutation), CMT2A (MFN2 mutation), CMT1B (MPZ mutation) and HNPP (PMP22 deletion; table 1). These five subtypes of CMT accounted for 89.2% of all of the genetically confirmed mutations. The remaining subtypes accounted for <1% each, with the exception of HSN1, which accounted for 2.5% of patients with a known genetic diagnosis (table 1). The relatively high number of patients with HSN1 results from the large number of patients enrolled at a single centre (National Hospital for Neurology and Neurosurgery, UK). CMT1A comprised 67.5% of patients with a known genetic diagnosis and 36.9% of total patients. Among patients with CMT2, 29.5% had CMT2A. Three patients had mutations in multiple genes.
Frequency of CMT subtypes
Patient impairment
Baseline CMTNS (table 2, see online supplementary tables S1 and S2) and CMTES were available for 906 and 1316 patients, respectively. The mean CMTNS for CMT1A was 13.7, which is 38% of the total score of 36 points, and is similar to what has been reported in other series.4–6 The mean CMTES for CMT1A was 9, which is 32% of the total score of 28. The mean CMTNS for all patients with CMT1X was 13, and for male patients only was 15.9. This is in the range of 11–16 as previously reported for males in their second and third decades of life.7 However, there were some unexpected findings for patients with CMT1B and CMT2A, two of the other common subtypes of CMT. The mean CMTNS for CMT2A—14.3—was much lower than the value of 21 reported previously.8 The mean and SD (13.7, SD 7.7) of the CMTNS for CMT1B were similar to those of CMT1A (13.7, SD 6.5). Previous reports of CMT1B have emphasised phenotypic differences in childhood versus adult onset forms of the disease such that we would have predicted a much wider SD in CMT1B even if their mean CMTNS were similar.9 ,10
CMTNS for common mutations
A newer version of the CMTNS, CMTNSv2, was developed and validated in 2011 to make the CMTNS more sensitive to change over time. We compared the CMTNS and CMTNSv2 baseline values in patients with CMT1A, who comprised our largest subtype and have an identical genetic cause (the duplication of PMP22).11 ,12 The mean/SD was 14.7/3.6 (n=54) and 13.5/3.6 (n=271) for CMTNS versions 1 and 2, respectively. The mean/SD was 9/2.8 (n=53) and 8.5/2.8 (n=462) for CMTES versions 1 and 2, respectively.
CMT impairment is known to increase with age.13 This is also evident in our patients with mean CMTNS increasing by 0.124 points for every additional year of age. To determine whether the age of the patients we evaluated was affecting our baseline scores, we evaluated the mean ages of patients with the four most common subtypes. The mean ages/SD for CMT1A (35/20), CMT1B (40/20), CMT2A (32/20) and CMT1X (39/17) were similar. We additionally adjusted the mean CMTNS for age and gender and found that while both variables were statistically significant, the mean CMTNS were not significantly altered nor the variance in scores significantly decreased for the common mutations (see online supplementary table S3). The differences between the CMTNS and CMTES of different CMT subtypes are therefore unlikely to be due to the age of the patients.
We next investigated whether different sites obtained similar CMTNS and CMTES for patients with the most common subtypes—CMT1A, CMT1B, CMT1X and CMT2A (table 3 and see online supplementary table S4). We compared the results from each of the four sites that had evaluated the largest numbers of patients, and pooled results from the remaining sites to minimise the effects from individual sites that had evaluated fewer patients. A statistically significant intersite variability in CMTNS was detected among sites for the common subtypes of CMT (p<0.0001). This significant difference was completely eliminated, however, by excluding the Milan site. These results were not altered by adjusting for age and gender (see online supplementary table S3).
Mean CTMNS for individual sites
Rare forms of CMT
To date, we have collected data on 78 patients with rarer forms of CMT, including subtypes of CMT1, CMT2 and CMT4 (here considered to be all recessive forms), HMN and HSN. Specific characteristics of these patients are provided in online supplementary tables S1 and S2.
Discussion
Genetic distribution of CMT in the INC
Our data are from over 1650 patients with various forms of CMT; the largest collection of patients with CMT reported to date. As the data come from multiple sites, we compared our results with what has been previously reported from single sites or smaller series (see online supplementary tables S5 and S6). These comparisons must be interpreted cautiously, however, as the data presented in online supplementary table S5 include populations from varied geographical regions. Furthermore, the way in which CMT was defined and classified differed between the various series. The largest single centre studies of CMT include Detroit,10 London,14 France15 and Germany.16 In all centres, CMT1A was, by far, the most common form of CMT, followed by CMT1X, HNPP, CMT1B and CMT2A. These five diagnoses cumulatively accounted for 89% of all genetic diagnoses in our series, similar to previous studies (91–94%). The remaining genes tested accounted for less than 2.4% of each of the positive genetic diagnoses. Many of the previously published data come from single centres in the USA, the UK and northern Europe.10 ,14–17 Since most INC sites are located within these regions and many American and Australian ancestors are from Northern Europe, it is not surprising that our results are similar. The distribution of subtypes, varies in different geographical regions, such as Greece,18 Norway,19 Spain20 and Japan,21 and may differ even more where autosomal recessive forms of CMT are more common22 (see online supplementary tables S5 and S6).
In our current series, 60.4% of patients enrolled obtained a genetic diagnosis, a similar value to the 54–67% observed in other large series from the US and Northern Europe (see online supplementary table S5). Our results were also similar to those of previously published studies in that CMT1 was more common in our patient population than CMT2 and had higher genetic testing success rates (91% of patients with CMT1 received a genetic diagnosis versus only 43% of patients with CMT2). The success rate for genetic diagnosis in CMT1 was higher than that reported by the London group (84%)23 and lower than that in the Detroit series (98%).10 The variation in these values most likely reflects a referral bias, as some centres may selectively see more patients in whom common mutations have already been excluded. Our diagnostic hit rates were also higher than those in which patients were not evaluated in CMT centres,14 ,16 ,24 confirming that making a genetic diagnosis is more likely for patients evaluated in centres with expertise in CMT.
CMTNS and outcome measures
A unique feature of our data is that it includes clinical outcome measures such as the CMTNS, CMTNSv2 and CMTES for all patients. Thus, we are recording the frequency of different genotypes as well as recording quantified impairment scores for all patients in a standardised fashion. We believe that these data are useful not only in defining the severity of each subtype but also as a baseline for longitudinal natural history studies of various subtypes of CMT. With regard to the specific outcome measures, the CMTNS3 and the CMTNSv223 have been validated, and both are the currently suggested outcome measures for CMT by the National Institutes of Neurological Diseases and Stroke (NINDS) Common Data Elements resource guide. The CMTNS has been shown to correlate well with other measures of disability including the ambulation index, self-assessment and hand function questionnaires, the Nine-Hole Peg Test, and the neuropathy impairment score.3 Nevertheless, we recognise that there are potential limitations for the CMTNS and CMTNSv2 in natural history investigations. Recent Italian/UK5 and American6 clinical trials of ascorbic acid treatment for CMT1A were not able to detect significant progression with the CMTNS over a 2-year period. Although the CMTNSv2 has been designed to be more sensitive to change than the CMTNS,23 and has subsequently undergone Rasch analysis to further improve its sensitivity,25 it has not yet been tested in longitudinal studies. The INC is continuing to develop outcome measures including a paediatric instrument, the CMT Pediatric Scale (CMTPedS),26 patient-reported disability score,27 CMT-specific quality of life instruments for adults and children, and impairment scores for infants. We expect data from these measures to be increasingly available within the INC such that investigators will be able to compare these instruments with the CMTNS in future studies.
Our findings did show a significant intersite variability in the CMTNS; however, this variability was largely eliminated by excluding patients from one centre where patients had received lower scores. Investigators in the INC have been cross-trained on common patients, have taken online training courses in the performance of the CMTNS, and use standardised questioning and examination techniques to ensure that evaluations of patients at one centre will be similar to those at other sites. Inter-rater variability has also been evaluated, showing a correlation coefficient between the total CMTNS for two examiners of 0.98 for CMTNS and 0.97 for CMTNv2 (p<0.01).3 ,23 We therefore suspect that the difference in scoring at the Milan site reflects the still low number of patients recruited, allowing for large families with mild phenotypes to have a disproportionate impact on the findings. Nevertheless, the differences between sites also emphasise the need for continued training to ensure that differences in methodology do not unduly influence the natural history studies.
Site-specific scoring
The overall CMTNS, CMTNSv2 and CMTES from INC sites for CMT1A were similar to what has been reported from single centres, which suggests that sites were evaluating a representative group of patients.4–6 Similarly, scores for CMT1X were similar to what has been reported.7 The scores for CMT1B and CMT2A, however, differed from prior studies. Both the mean CMTNS for CMT1B and SD of 7.7 were similar to those of CMT1A (13.7 and 6.5). Previous reports on CMT1B have emphasised phenotypic differences between early and adult onset forms of disease, such that we would have predicted a much wider SD in patients with CMT1B even if their mean CMTNS were similar.9 ,10 The mean CMTNS for CMT2A of 14.3 was similar to that for CMT1A and much lower than the value of 21 reported previously.8 The differences in CMT1A and CMT1X compared with CMT1B and CMT2A are probably related, at least in part, to differences in their pathogenesis. Mutations in CMT1A are all caused by the same duplication of PMP22 on chromosome 1711 ,12 such that most patients have a similar phenotype.28 More than 400 reported GJB1 mutations that cause CMT1X have been reported.29 Affected males with both point mutations and deletions in the GJB1 gene demonstrate a similar, age-related phenotype, suggesting a loss of function mechanism of disease.7 Therefore, most male patients with CMT1X would be predicted to have a CMTNS similar to what has been previously reported. In contrast, the phenotypes of both CMT1B and CMT2A are known to vary depending on the particular mutation. Patients with CMT1B typically have a severe neuropathy with onset in infancy or an onset in adulthood that may be much milder.9 ,10 Most MFN2 disease causing mutations cause a severe neuropathy in which patients become wheelchair users by young adulthood.8 The current study did not include young children who are assessed using the CMTPedS, and it is likely that participants with the most severe phenotypes were therefore under-represented. In addition, there may be variability in the distribution of patients in specific clinics that affects our overall scores. This is particularly likely as the total number of patients with CMT1B and CMT2A was 67 and 70 patients, respectively, and when this is divided into 13 centres, it is possible that any 1 centre may be enrolling a number of patients from an individual family rather than multiple index cases. For example in the CMT1B cohort, the mean CMTNS in the London site was 19.8 compared with the Detroit site of 12. In the London site, most patients were enrolled from a large individual family with very severe early onset CMT1B. As the enrolment of these two subtypes increases, we may get a better picture of the true impairment in these two subtypes of CMT. There is also the added complication in CMT2A that it is sometimes difficult to be sure a mutation is pathogenic using current validation techniques. As we improve our ability to validate pathogenic mutations, we may find that some of the patients with milder CMT2A harbour mutations in different genes and this may have skewed the CMTNS.
Challenges with rare forms of CMT
A major goal of the INC is to increase our enrolment of patients who have rare forms of CMT, so that we can generate natural history data. We collected data on 78 patients with rare forms of CMT, but this breaks down into just a few patients per genotype. This is a good start, but we need to evaluate more patients to define the natural history of the rare forms of CMT. To capture this group of patients, we need to obtain data from regions such as North Africa where up to 40% of patients with CMT are reported to have autosomal recessive inheritance.22
Acknowledgments
This INC is a part of the NIH Rare Diseases Clinical Research Network (RDCRN). The authors would like to thank all the patients who participated in the INC and their families. Without whom this study would not be possible. They would also like to thank the people working at INC sites who contributed to this study. These include Julian Blake, Betsy Burgos, Daniela Calabrese, Vinay Chaudhry, David Cornblath, Katy Eichinger, Tim Estilow, Claudia Gandioli, Audra Hamilton, Allan M Glanzman, Ahmet Hoke, Andrea Kelley, Livija Medne, Manoj Menezes, Sinead Murphy, Jillian Olsen, Giuseppe Piscosquito, Alex Rossor, Oranee Sanmaneechai, Anna Sorey, Mariola Skorupinska, Janet Sowden and Andrea Swenson.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors VF wrote the first draft of the manuscript and collected and analysed data. BB performed statistical analysis for the study. MMR and MES were involved in the study conception, collected and analysed data and edited drafts of the manuscript. CAK managed the data and edited drafts of the manuscript. SZ contributed to genetic analysis for this manuscript. SSS collected data, helped edit drafts of the manuscript and contributed tables to the manuscript. DP collected and analysed data and edited drafts of the manuscript. JB, JD, RSF, DNH and ML collected data and edited drafts of the manuscript. CB, SF, TG, JL, TL, CJS, FM, GP, SR, RS, CES, SWY, IM and EP collected data.
Funding Funding and/or programmatic support for this project has been provided by U54NS065712 from the National Institutes of Neurological Diseases and Stroke (NINDS) and the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Sciences (NCATS). The INC also receives support from the Muscular Dystrophy Association (MDA) and the Charcot Marie Tooth Association (CMTA). Additional support for this study includes the NINDS grant R01NS066927 and Vanderbilt Institute for Clinical and Translational Research fund VR1687 to JL. Part of this work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
Competing interests None.
Ethics approval University of Iowa IRB and Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement VF had full access to the data, and if requested, will provide access to the data on which the manuscript was based to the editors.