Article Text

Abstract
Induced pluripotent stem cells (iPSCs), which greatly circumvent the ethical issue of human embryonic stem cells (ESCs), can be induced to differentiate to dopaminergic (DA) neurons, and hence be used as a human disease model for Parkinson's disease (PD). iPSCs can be also utilised to probe the mechanism, and serve as an ‘in vivo’ platform for drug screening and for cell-replacement therapies. However, any clinical trial approaches should be extensively supported by validated robust biological evidence (based on previous experience with fetal mesencephalic transplantation), in particular, the production and selection of the ‘ideal’ neurons (functional units with no oncological risk), together with the careful screening of appropriate candidates (such as genetic carriers), with inbuilt safeguards (safety studies) in the evaluation and monitoring (functional neuroimaging of both DA and non-DA system) of trial subjects. While iPSCs hold great promise for PD, there are still numerous scientific and clinical challenges that need to be surmounted before any clinical application can be safely introduced.
- PARKINSON'S DISEASE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Parkinson's disease (PD), a common neurodegenerative disorder, is characterised by a selective loss of dopaminergic (DA) neurons from the pars compacta of the substantia nigra, and the formation of Lewy bodies. While tremor, bradykinesia, rigidity and postural instability are common signs and symptoms, non-DA features such as gait disturbance and dementia may occur when non-DA systems are involved, and these can be clinically disabling and difficult to treat.1
Research on PD has focused on dissecting the pathophysiological mechanism in order to develop novel therapeutic strategies. Mitochondrial dysfunction, oxidative stress, kinase pathways, calcium dysregulation and inflammation, are implicated in PD.1 Numerous cellular and animal models have been established to unravel the mechanism behind PD. However, the discoveries garnered from cellular models, which are usually not based on human DA neurons, may not fully reflect the pathogenesis in DA neurons with PD.2 In addition, animals may have different vulnerability to the disease-causing factors. Therefore, stressors or genetic risk factors, which play an important role in the pathogenesis of PD in human, may not be revealed by animal studies.3
Induced pluripotent stem cells (iPSCs) refer to a group of pluripotent stem cells that can be generated from adult cells. iPSCs share a crucial property with embryonic stem cells (ESCs) as they have a potency to differentiate into any tissue in the body. Moreover, various terminally differentiated cells can be utilised to generate iPSCs, avoiding destruction of an embryo entailed by the generation of ESCs. Thus, iPSCs hold great promise for regenerative medicine, and PD is the prototype neurodegenerative disease that will most likely benefit from this scientific breakthrough since the disease affects the substantia nigra predominantly in the early stages.
In this review, we provide an update on recent progress and potential limitations on the use of iPSCs in PD research, and, in particular, on how iPSCs can help facilitate the understanding of the pathophysiology and the development of therapeutics in PD (figure 1).

Outline of applications of induced pluripotent stem cells (iPSCs) on Parkinson's disease (PD). iPSCs generated from patients with PD may be utilised in research as well as in clinical application. iPSCs can further differentiate to dopaminergic neurons and model PD for the exploration of mechanism and search for therapeutic compounds. Clinically, iPSC-derived neurons may facilitate PD diagnosis by reflecting pathogenesis in the brain. Furthermore, iPSC-derived neurons also hold attractive potential in cell therapy for PD.
Discovery of iPSCs
In 2006, Dr Yamanaka's team discovered that a cocktail of four transcription factors, that is, OCT4, SOX2, KLF4 and c-Myc, could reprogramme skin fibroblasts to pluripotent cells. These cells were able to differentiate into various types of cells from the three primary germ layers, which represent key attributes of ESCs.4 However, the biological properties of the reprogrammed cells differed from ESCs. For instance, we have previously demonstrated that to maintain self-renewal of the pluripotent cells, Oct4 and Nanog are bound to their own promoters to form an autoregulatory loop, which was not established in the reprogrammed cells.5 In addition, injection of iPSCs into blastocysts failed to generate a viable chimaeric embryo.4 Efforts have since been made to find the optimal way for reprogramming—and to improve the quality of the iPSCs—so that the reprogrammed cells can maximally resemble the ESCs in terms of their biological features.
Source of iPSCs
Researchers have been experimenting with various cell sources for reprogramming. Today, iPSCs can be generated from different origins, such as liver and stomach cells, neural stem cells and peripheral blood cells.6 ,7 The success of peripheral blood cells that act as templates for reprogramming attracts clinical interest as drawing blood is the least invasive operation and rarely presents complications. We were involved in further optimising the protocol and made it possible to use as little as 10 µL of finger tip capillary blood to reprogramme blood cells to iPSCs.6 Another interesting technological breakthrough of iPSCs is that keratinocytes from hair follicles were successfully reprogrammed to iPSCs,8 though the amount of hair that has to be plucked might be unacceptable to donors. We can foresee that, to further improve accessibility, it may be possible, in the future, to generate iPSCs with a mouth swab.
Concern of tumourigenesis
The risk of tumourigenesis has always weighed on the minds of researchers. This concern stems from the known oncogenes utilised in the reprogramming of iPSCs, including Myc and Klf4. Other genes involved in generation of iPSCs, Nanog, Sox2 and Oct4, are also linked to tumourigenesis.9 Of note, a canonical tumour suppressor, p53, is closely related to the generation of iPSCs. It was found that elevated p53 levels may significantly attenuate reprogramming through DNA damage response and p53-dependent apoptosis.10 Another reason for iPSC-related tumourigenesis lies in the disruption of the genome integrity. This could be caused either by virus-mediated integration or by in vivo somatic mutations.11 In order to retain genome integrity throughout the reprogramming, various techniques have been developed to generate insertion-free iPSCs, such as synthetic modified mRNA, episomal vectors, Sendai virus vectors and others.7 However, even if there is no damage in genome caused by retroviral insertion, genome integrity is still a concern during reprogramming. Gore et al11 reported that, in iPSCs that were reprogrammed through diverse methods, including non-integrating methods, mutations of non-synonymous, non-sense or splice variants were discovered, with most of the mutations located in the genes related to cancers. Therefore, although great efforts have been made to reduce the risk of tumourigenesis, iPSC-derived neurons can be applied in clinical trials only after stringent selection, including extensive genetic screening for de novo mutations generated from the reprogramming and in vivo testing by transplantation of the neuron graft into a primate model.
Modelling PD using iPSCs
Unravelling mechanism
Since iPSCs can specifically differentiate to A9 DA neurons, the incomparable advantage of biological relevance makes iPSCs use an excellent system to model PD. iPSCs provide an opportunity to verify the understanding of PD harvested from cellular or animal models and pathological studies in live DA neurons. For instance, the most exciting finding made recently on PD mechanism is that mitophagy is implicated in PD. On mitochondrial depolarisation, PINK1, a mitochondrion-targeted kinase, accumulates on outer mitochondrial membrane, recruiting Parkin and initiating mitophagy. It is interesting that Parkin/PINK1-mediated mitophagy has been well demonstrated in the cancer-derived cell lines, while results of Parkin/PINK1-mediated mitophagy in neurons have been conflicting. For instance, Van Laar et al12 reported that, in rat neurons, mitochondrial depolarisation failed to induce Parkin translocation to mitochondria. In contrast, Cai et al13 demonstrated that mitochondrial depolarisation did induce Parkin translocation to mitochondria in mouse neurons, but at a much lower speed. However, as mentioned earlier, DA neurons from rodents might be distinct from human DA neurons. Therefore it is indispensable to verify the mitophagy in human DA neurons. In iPSC-derived neurons, it has been found that mutations in PINK1 did compromise Parkin translocation to mitochondria, eventually blocking autophagic clearance of mitochondria.14 However, endogenous Parkin was found not sufficient to mediate carbonyl cyanide 3-chlorophenylhydrazone (CCCP)-induced mitophagy in iPSC-derived neurons,15 which does not fit in the current model for the Parkin/PINK1-dependent mitophagy. We reason that CCCP may pose a PD-irrelevant scenario to cells by causing global mitochondrial depolarisation beyond rescue. Under the stress of CCCP, neurons in vivo might rather retain impaired mitochondria that may still provide energy than remove the whole mitochondrial system with the aid of exogenous Parkin. Therefore, establishment of restrained damage to a small amount of mitochondria in the iPSC-derived neurons should be crucial for the assessment of the role of Parkin/PINK1 in mitophagy and the significance of mitophagy in the pathogenesis of PD. Furthermore, since cells employ multiple layers of quality control to ensure their well-being, it is possible that defect in mitophagy only compromises cell viability in aged cells where the other protective machineries are either dysfunctional or overloaded. Recently, artificial ageing, including long-time culture16 and expression of progerin,17 was utilised to induce phenotypes in iPSC-derived neurons to reproduce features of PD, though the effects of the artificial ageing on mitophagy are yet to be studied.
Therapeutics
iPSC-derived PD models can be used for screening therapeutic compounds. Coenzyme Q10, rapamycin and the LRRK2 kinase inhibitor GW5074, have been demonstrated to rescue cytotoxicity caused by valinomycin or concanamycin A in iPSC-derived neurons from patients with PD. Furthermore, the finding that rapamycin and GW5074 selectively reduced reactive oxygen species production in iPSC-derived neurons with PINK1 mutation but not in neural cells from healthy subjects, highlights the difference in susceptibility to pharmaceutical compounds between diseased neurons and artificial disease models.18 More recently, nitrosative/oxidative stress was found to cause mitochondrial dysfunction and apoptotic cell death in A9 DA neurons with A53T α-synuclein mutation through S-nitrosylation of transcription factor MEF2C, which could be well targeted by a small molecule, isoxazole.19 Therefore, iPSC-based technology may facilitate identification of therapeutic compounds by elucidating authentic signalling pathways in diseased human neurons rather than artificial models. In addition, typical pathological features, such as Lewy bodies, identified in iPSC-derived neurons, may have diagnostic and therapeutic potential. For instance, correction of the A53T α-synuclein mutation by gene editing eliminates the Lewy bodies with phosphorylated α-synuclein in iPSC-derived neurons.19 Hence identification of a unique PD pathological hallmark in iPSC-derived neurons can provide a specific and relevant readout for drug development compared with other cellular events, such as cell death.
Drug screening on iPSC-derived neurons could be implemented in two ways. First, the drug candidates could be tested in vivo on the iPSC-derived neurons generated from the patient who needs the medication. If the drug could improve pathophysiological features in the neurons, it should be effective for the same patient. Although drug screening in specific patient iPSC-derived neurons provides an appealing perspective for the individual, it is time-consuming and may not be affordable in the near future. The other way is to test the drugs in iPSC-derived neurons with different PD genotypes. Genome-wide association studies (GWAS) have revealed a number of gene variants that modulate the risk of PD. For instance, a recent meta-analysis of GWAS data identified 28 independent risk variants for PD across 24 loci.20 With the rapid development of genome sequencing, it would be very convenient to get the genome information of the patient and categorise the patient to a certain group defined by its PD-related genotype. If we are able to complete drug screening among these subgroups, a list of customised drugs suitable for the patient's genotype can be ready once the disease is diagnosed.
Clinical applications
Diagnostic
A recent paper on clinicopathological correlations of PD highlighted the need to develop new methods to improve diagnostic accuracy of early PD.21 iPSCs derived from patients may serve this need if the phenotypes shown in iPSC-derived neurons can recapitulate the pathological features in PD, including selective loss of DA neurons and formation of Lewy bodies. Indeed, cell death has been found in most iPSC-derived neurons from patients under stressors17 ,19 ,22 or over prolonged culture.16 However, the results for the occurrence of Lewy bodies remain inconsistent. Lewy bodies and α-synuclein aggregation were identified in A9 DA neurons derived from iPSCs with A53 T α-synuclein mutation, but not in the corrected isogenic controls.19 In another study, expression of progerin was used to induce ageing in PD iPSC-derived DA neurons, where characteristic phenotypes were revealed, though the Lewy body precursor inclusions were found only in iPSC-derived DA neurons with Parkin V324A mutation.17 The presence of Lewy body precursor inclusion in the neurons carrying Parkin mutation is interesting, as patients harbouring Parkin mutation usually do not have Lewy bodies. It is unknown if the α-synuclein aggregates found in these studies are of pathogenic traits (such as filamentous structure). The conformation of α-synuclein strain has recently been found to dictate its impact on histopathological and behavioural phenotypes, with α-synuclein inclusion possessing maximal neurotoxicity in the form of fibrils, while ribbons leading to the phenocopy of PD.23 In contrast, though DA neurons derived from iPSCs with LRRK2 G2019S mutation showed either upregulation of α-synuclein protein,22 or diffuse cytoplasmic accumulations of α-synuclein,16 no obvious aggregates or inclusions were reported.16 ,22 Of note, a duration of 75 days of culture of DA neurons was used to mimic ageing and to promote development of PD-related phenotypes.16 However, the optimal culture duration of differentiated neurons to mimic PD is unclear. In addition, progerin is responsible for Hutchinson-Gilford progeria syndrome, a disease of premature ageing, where the nervous system is not compromised. Therefore, the characteristic PD-related phenotypes might not be reproduced in iPSC-derived neurons with progerin-induced ageing. In summary, taking into account the inconsistencies in the findings of α-synuclein aggregates, iPSC technology needs to be optimised and standardised before meaningful inferences can be drawn.
Careful examination of iPSC-derived neurons may reveal subtle changes preceding the formation of pathological lesions, including mitochondrial dysfunction, oxidative stress, calcium dysregulation and other cellular events compromised in PD,1 which may act as biomarkers for the early diagnostic of PD. However, these early changes, such as mitochondrial dysfunction and disturbance in autophagy, are present not only in PD but are also shared by some other neurodegenerative diseases. In addition, diverse detrimental factors trigger distinct cascades, but result in similar damage to the vulnerable DA neurons, eventually leading to PD. Therefore, some pathophysiological changes, especially those occurring in the upstream of the pathway, might only show up in a certain group of PD and not be shared by all PD cases. Identification of a group of biochemical markers with the aid of iPSC technology and determining the right combination of these changes can potentially contribute to diagnostic accuracy in early PD. However, it is premature at this moment to draw conclusions without identification of consistent and specific marker(s).
Cellular replacement
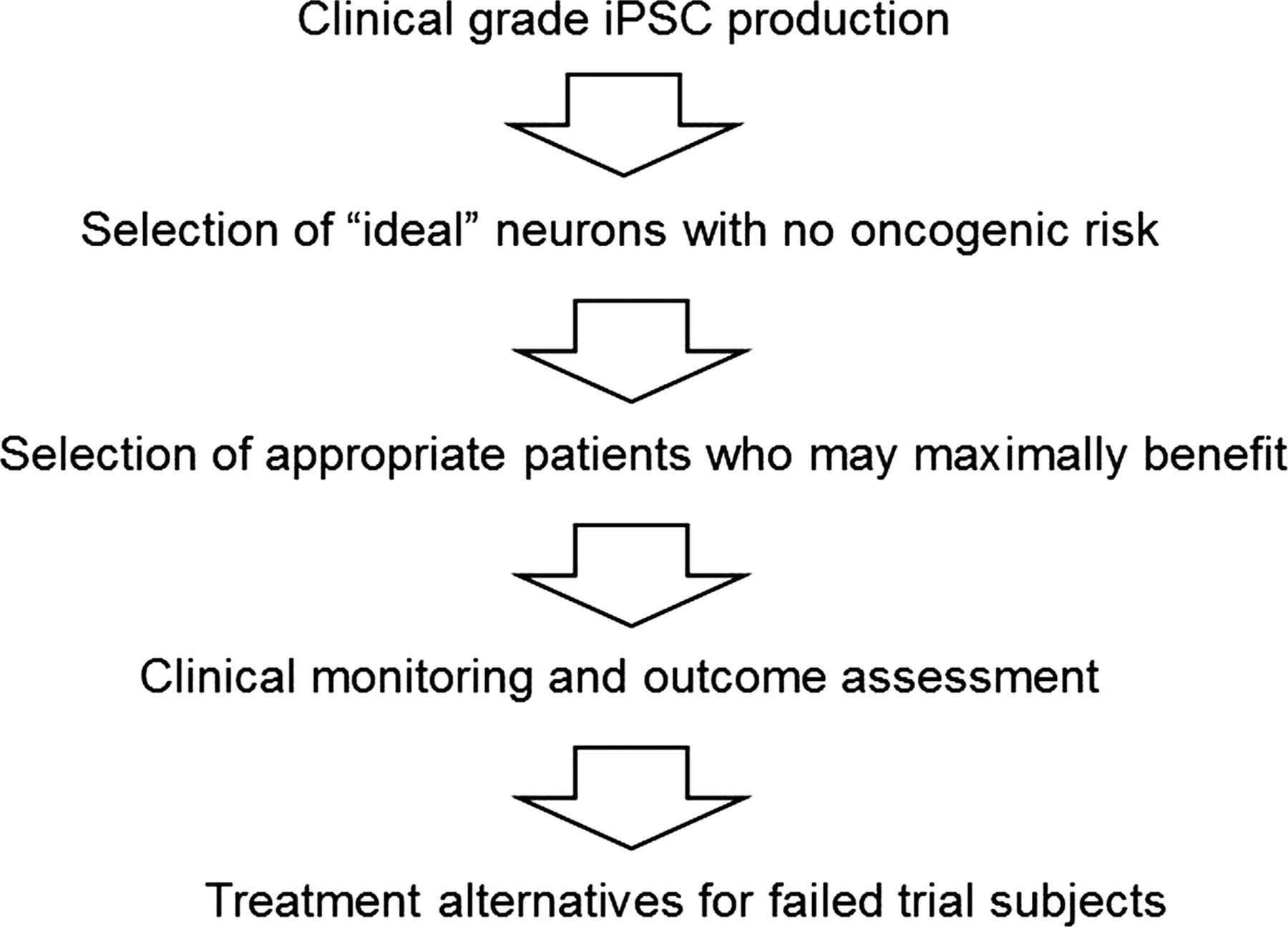
Unlike injury to the central nervous system, where subsequent glial proliferation eventually sequesters the lesion, grafted neurons in PD may have a better chance of compensating the function of diminished neurons without the barrier of glial scarring. Indeed, inspiring success in PD treatment has been achieved through allograft of human embryonic mesencephalic tissue.24 Recent attempts to transplant neural progenitor cells (NPCs) or DA neurons from ESCs in rodent and primate PD models have also yielded encouraging results.25 However, embryonic or fetal neural transplantation procedures are limited by availability of tissues, potential immunorejection and ethical concerns. Fortunately, iPSCs have unlimited proliferative capacity and are hence able to provide an inexhaustible supply of neurons for cell replacement. This also obviates ethical concerns such as those with regard to fetal tissues. A pioneering study showed that grafted iPSCs were able to differentiate into DA neurons and rescue motor deficits in a rat PD model.26 Subsequently, in a primate model, DA neurons were reported to differentiate from human iPSC-derived NPCs after graft. The DA neurons survived for up to 6 months, though the improvement in behaviour was not statistically significant.27 In addition to investigating iPSC-based cell therapy in different model system, iPSCs generated through distinct approaches have been utilised to optimise the therapy. Protein-based iPSC-induced NPCs were reported to differentiate to the DA neurons after transplantation, and to overcome the early senescence and apoptosis caused by virus-based reprogramming.28 These results provide a favourable outlook for the application of transplantation of iPSCs to the treatment of PD. However, a stringent clinical study paradigm is advisable for iPSC-based cellular therapy trials (figure 2).
{kind=link}
{kind=link}
Clinical trial paradigm (iPSC, induced pluripotent stem cells).
Preparation of graft cells
Cell therapy requires optimal grafting cells, as the properties of the grafting cells, such as their purity, number and stage, have significant impact on the efficacy and safety of transplantation. The lesions from fetal neuronal grafts have shown us the importance of the purity of the graft. The heterogeneity of harvested embryonic mesencephalic tissue has been considered the major reason for huge variability of improvement in motor defects in previous studies. Moreover, dyskinesia, a major adverse effect of fetal neural grafts, has been found to be mediated by serotonergic neurons.29 In contrast, optimised protocol for differentiating ESCs yields bona fide A9 DA cells in high percentage, dramatically restoring motor function.25 In addition, a study showed that DA neuron grafts sorted using a NCAM+/CD29low marker gave rise to better survival of DA neurons and better function recovery compared to unsorted grafts.30 Tumourigenesis might be addressed with grafts of high purity, as few proliferating cells were detected in the grafts in 6-hydroxydopamine-challenged rats, using cell sorting with a floor plate marker, CORIN.31 In terms of cell number, though 100 000 DA neurons have been proposed for a successful graft, neural outgrowth was observed in a high concentration of NPC grafts.28 Mid-stage ESC-derived DA neurons have been shown to have better survival and greater rescue of motor defects than neurons in other stages.32 Moreover, the way to induce differentiation of iPSCs is also crucial for ideal replenishment of DA neurons, as rosette-derived grafts resulted in much more robust proliferation than did floor plate-based strategy derived grafts.25 Since iPSCs are prone to tumourigenesis during both reprogramming and differentiating stages, the aforementioned genetic test and in vivo screening must be performed to secure the production of clinical grade iPSCs.
It is also important to determine whether the iPSC graft is sourced from the individual patient or from a HLA-matched healthy donor. Undoubtedly, the neurons derived from the patient should have the best immune compatibility for transplantation. However, there are still key issues that have to be taken into account for the autotransplantation. First, if the genetic defects in the patient's cells from which the iPSCs are generated are not corrected, the transplantation of the iPSC-derived neurons or NPCs would predispose the patient to neurodegeneration. Even though gene correction can currently be achieved through genome-editing techniques, such as the clustered regularly interspaced short palindromic repeats-Cas9 system and transcription activator–like effector nucleases, fully rectifying PD-related variations is still impractical, as it is the cumulative effect of dozens of variations that lead to PD.20 Furthermore, a number of risk variants might have been skipped due to inadequate sample size of subjects or might yet be identified by contemporary genomic approaches. In contrast, it would be much more economical and practical if the patient is treated with a HLA-matched neuron graft from a tested global iPSC library.33
Patient selection
It is noteworthy that a patient who received transplants of embryonic mesencephalic tissue experienced a significant initial improvement, followed by gradual deterioration with refractory non-DA features, including difficulty in gait and balance, and falling, indicating DA neuron replacement fails to reverse the progression of PD to a non-DA system.34 Along with the observation of exclusive survival of DA neurons in animal models after iPSC-induced NPC graft, we reason that DA neuron/NPC grafts may have limited effect on advanced disease that develops beyond the DA system. A favourable transplant outcome predicted by a good preoperative response to levodopa also supports this point.35 Therefore, we propose that the risk of involvement of a non-DA system should be evaluated using positron emission tomography or other structural and functional imaging to better predict the benefit of receiving a DA neuron/NPC graft for a particular patient. Moreover, patients with genetic PD, especially those with mutation in parkin, usually experience a slower disease progression and suffer less non-motor symptoms,36 which indicate relatively exclusive DA involvement. Therefore, patients with genetic PD may possibly expect a better outcome of cell therapy in terms of long-term effect.
Microenvironment in PD
Since the ultimate goal of PD therapy is to halt disease progression by rescuing the underling pathophysiological damage in the brain, cell therapy may never over-ride DA replacement by drugs if surviving grafted neurons cannot provide more benefit than mere replenishment of diminished DA neurons. In contrast to improving the catastrophic microenvironment by graft, it has been found that the microenvironment around the graft site negatively affects the graft. Host-to-graft disease propagation has been identified in patients with PD with development of Lewy bodies in grafted neurons.37 Subsequent studies revealed that intercellular transmission of α-synuclein from host-to-graft might contribute to disease propagation by promoting α-synuclein aggregation in recipient neurons.38 The Prion hypothesis has since been brought up to highlight the propensity of synuclein aggregates to disseminate through the nervous system. Although PD is not a contagious disease, α-synuclein shares common features with prion protein. Once acquiring β-sheet conformation, aggregated α-synuclein is able to induce Lewy pathology by entrapping soluble α-synuclein. Subsequently, mature aggregates may split into small fragments, which may act as seeds of new aggregates and transmit to adjacent cells.39 Therefore, grafted neurons are susceptible to a vicious cycle of aggregation formation and spreading of the aggregates among neurons. Moreover, though favourable growth of DA neurons has been found in a neurotoxin-lesioned animal model, it is uncertain how well DA neurons would be able to survive in a patient's brain, as pulse treatment of neurotoxin is usually discontinued while the adverse milieu in PD remains. Host-to-graft disease propagation and persistent PD milieu highlight the importance of improving the adverse microenvironment of PD, which in turn requires the elucidation of the mechanism of PD with the aid of the iPSC PD model. Fortunately, the impact between host microenvironment and graft cells is not unidirectional. Redmond et al40 reported that a much larger number of astrocytes were derived from grafted human NSC and express neuronal protective factors, thereby adjusting homeostasis of the microenvironment. Owing to variable microenvironments in different patients and limited function of sheer DA neurons to improve adverse pathogenesis, constructing a functionally autonomous DA ‘unit’ might be a future direction to develop graft candidates possibly with certain growth factors or even glia cells in addition to DA neurons. The comprehensive DA ‘unit’ may provide not only the replacement of DA neurons, but also elements that may improve the adverse microenvironment in patients with PD. Moreover, the autonomy of the graft is particularly important in terms of controlling of the graft cell numbers, as the grafted cells are more directly exposed to the artificial microenvironment in the ‘unit’ than variable surrounding milieu, which may affect the ideal in vitro protocol.
Conclusions
iPSC-derived neurons provide an exciting system for PD modelling, which can be utilised for the investigation of disease pathogenesis, with the potential for development as a clinical diagnostic tool, and also offer new therapeutics for patients (table 1). However, while iPSC-derived neurons have enormous potential in PD, any clinical trial approaches should be extensively supported by validated robust biological evidence (based on previous experience with fetal mesencephalic transplantation), in particular, the production and selection of ‘ideal’ neurons (functional units with no oncological risk), together with the careful screening of appropriate candidates (such as genetic carriers) with inbuilt safeguards (prior safety studies) in the evaluation and monitoring (such as functional neuroimaging of both DA and non-DA system) of trial subjects. We wish to believe, with cautious optimism, that some of these enormous challenges can be surmounted in the near future.
Current approaches and future directions in the application of iPSCs
Acknowledgments
This study was supported by the National Medical Research Council (STaR award and Parkinson's disease clinical translational research programme).
References
Footnotes
Contributors E-KT and BX planned the outline of the review. RT and HHN contributed ideas and suggestions. All the authors approved the manuscript.
Funding National Medical Research Council (grant number PD TCR).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.