Article Text

Abstract
Objective Antibodies to cell surface central nervous system proteins help to diagnose conditions which often respond to immunotherapies. The assessment of antibody assays needs to reflect their clinical utility. We report the results of a multicentre study of aquaporin (AQP) 4 antibody (AQP4-Ab) assays in neuromyelitis optica spectrum disorders (NMOSD).
Methods Coded samples from patients with neuromyelitis optica (NMO) or NMOSD (101) and controls (92) were tested at 15 European diagnostic centres using 21 assays including live (n=3) or fixed cell-based assays (n=10), flow cytometry (n=4), immunohistochemistry (n=3) and ELISA (n=1).
Results Results of tests on 92 controls identified 12assays as highly specific (0–1 false-positive results). 32 samples from 50 (64%) NMO sera and 34 from 51 (67%) NMOSD sera were positive on at least two of the 12 highly specific assays, leaving 35 patients with seronegative NMO/spectrum disorder (SD). On the basis of a combination of clinical phenotype and the highly specific assays, 66 AQP4-Ab seropositive samples were used to establish the sensitivities (51.5–100%) of all 21 assays. The specificities (85.8–100%) were based on 92 control samples and 35 seronegative NMO/SD patient samples.
Conclusions The cell-based assays were most sensitive and specific overall, but immunohistochemistry or flow cytometry could be equally accurate in specialist centres. Since patients with AQP4-Ab negative NMO/SD require different management, the use of both appropriate control samples and defined seronegative NMOSD samples is essential to evaluate these assays in a clinically meaningful way. The process described here can be applied to the evaluation of other antibody assays in the newly evolving field of autoimmune neurology.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Assays to detect pathogenic antibodies have gained importance in the past 10 years with the discovery of new diseases that appear to be mediated by antibodies to proteins such as aquaporin (AQP) 4 (identified in 2005),1 ,2 n-methyl-d-aspartate (NMDA) receptor (2007),3 ,4 glycine receptor (2008),5 a-amino-3-hydroxy-5-methyl-isoxazolepropionic acid receptor (2009),6 gamma-aminobutyric acid (GABA)B receptor (2009),7 leucine-rich, glioma inactivated 1 (LGI1) (2010),8 ,9 contactin-associated protein like 2 (CASPR2) (2010)9 ,10 and GABAA receptor (2014)11 ,12 among others (for reviews, see).13–15 The accurate and rapid detection of these antibodies in patient serum or cerebrospinal fluid (CSF) can lead to immunotherapies that reduce patient morbidity and mortality.
Neuromyelitis optica (NMO) was the first antibody-mediated central nervous system (CNS) disease with a clearly defined target, AQP4, identified in a variable proportion of patients. NMO can be defined clinically; the patients present with episodes of optic neuritis (ON) and transverse myelitis (TM) and brain lesions distinct from those found in multiple sclerosis (MS),16 but it is unusual for the full spectrum to be evident at the first episode. In the past, many patients with NMO have been misdiagnosed and treated with medications insufficient to control NMO disease activity such as interferon-β or natalizumab. With increasing use of AQP4-antibody tests, however, many patients with first episodes or partial syndromes of ON, myelitis or brainstem lesions have been reported with AQP4-antibodies. In these patients, a positive AQP4-antibody defines an NMO/NMO spectrum disorders (NMOSD) diagnosis, leading to prolonged immunotherapies with most likely reduced relapse rates. However, AQP4-antibody positivity differs widely between studies (33–90%), suggesting either poor sensitivities of some tests or false positives in patients with clinically definite NMO.17 ,18 False positives, which could lead to a diagnosis of NMO in patients with less requirement for aggressive/maintenance immunotherapies, have potential management implications.
Traditionally well-established and clinically defined patient groups are used to calculate sensitivities, but this is difficult when there are ‘seronegative’ patients, or patients who present with partial or atypical features. In this multicentre study, we compare AQP4 assay metrics on a mixed cohort of patient and control sera performed by 15 European centres that routinely test for AQP4-antibodies. We present a systematic approach that identifies assays that are most useful clinically. This process and results have implications for other antibody-mediated neurological disorders in this expanding area of clinical neurology.
Methods
Ethics
The research use of referred sera was approved by the Oxfordshire Research Ethics Committee ref 10/H0606/56, by the ethical review board of the University of Heidelberg, Germany, by the Regional and National Ethical Committee of Hungary (3893.316-12464/KK4/2010 and 42341-2/2013/EKU, Hungary), by the Ethics Committee of the Region of Southern Denmark (ref S-20120066), by the French data protection authority, by the regional committee for medical and health research ethics, Western Norway (REK#3.2006.1235), following Institutional Review Board (IRB) approval in Berlin, Dusseldorf and Munich, Germany, and according to the Dutch regulation for use of patient material.
Patient samples
All centres were asked to provide sera or plasma samples from 8 to 10 patients with AQP4-antibody positive or negative NMO or NMOSDs, excluding cases with unclear diagnoses or diagnoses complicated by related pathologies, and a similar number of clearly defined neurological control samples (eg, MS, other inflammatory neurological disease). Four groups provided samples only, whereas 15 groups performed assays only, and 6 groups provided samples and performed assays. A total of 209 coded sera/plasma samples were received by Euroimmun AG, Germany from 10 centres by May 2013 (16 were excluded due to insufficient volume, figure 1, table 1). The controls comprised samples from patients with a headache (39), MS (35 relapsing remitting, 2 primary progressive19), clinically isolated syndromes (4, all exhibited clinical and paraclinical features typical of MS), tumour (1 B-cell lymphoma, 1 colon carcinoma with neurological complications), Susac syndrome (1), progressive encephalomyelitis with rigidity and myoclonus (1), neuromyotonia (1), connective tissue disorder (1), myasthenia gravis (MG) (5) and acute disseminated encephalomyelitis (120). The test samples comprised 50 samples from patients who fulfilled the 2006 diagnostic criteria for NMO16 excluding AQP4 serostatus (35 submitted as seropositive from different centres based on their different AQP4 assays), and 51 samples were from patients with clinical features of NMO who did not meet the criteria (9 ON, 31 TM, and 11 with ON and TM; 39 submitted as seropositive by the centres; these were referred to as NMOSD in the context of this study). The NMO/spectrum disorder (SD) cohort was predominantly female (4.6:1) with a median age at sampling of 45 years and the samples were taken at a median of 3 years (range 0–30 years) from disease onset, mostly during remission (3·35:1).
Origin of samples

Study design. The coded clinical and serological data from 209 patients were sent to the University of Lübeck, Germany. The coded serum (199) or plasma samples (10) were sent to Euroimmun, Lübeck, Germany. Sixteen samples were excluded due to insufficient volume. In total, 193 samples were recoded, aliquoted (90–100 μL) and sent on dry ice to 15 European Centres to run 21 AQP4 assays with a 17-week deadline. Each centre entered its own data online to a web-based server maintained at the Institute of Quality Assurance, Lübeck, Germany. All clinical data, assay results and sample codes were sent to Oxford, UK and Lyon, France for initial analysis. A blinded overview was sent to each centre before unblinding the study in a meeting in Paris, France, where all groups were represented.
Study set-up and design
The clinical and paraclinical patient data were sent to the University of Lübeck, Germany (PT). The samples were stored at −20°C before recoding (EDEN001—EDEN193) and aliquoted into 90–100 μl samples. These were distributed on dry ice to the 15 participating centres, all of whom routinely test for AQP4-antibodies (see figure 1 for a flow diagram of the study design). A total of 17 weeks were allowed to perform the assay and results were entered online via a password-protected web-based interface established by the Institute for Quality Assurance, Lübeck, Germany (MP). If more than one method was used by a centre, results obtained from the different methods were entered separately. Centres could provide semiquantitative results only (negative, +, ++, +++ or ++++), or quantitative results. The semiquantitative data were converted to 0–4 to give a score for individual sera.
After completion of the assays, the clinical and paraclinical data from the University of Lübeck, a table of results from the Institute of Quality Assurance, a list of the original and newly assigned sample codes from Euroimmun, and all assay protocols were submitted to two evaluating groups (Oxford, UK and Lyon, France; figure 1). The data were analysed and a partially unblinded ‘overview’ figure (see online supplementary figure S1) was sent to all participants. All groups were represented at a meeting in Paris, France where this data set was fully unblinded.
Supplementary appendix 3
Assays
A total of 21 AQP4 assays were carried out (table 2): three live cell-based assays (CBA); 10 fixed commercial CBAs (Euroimmun AG, Lübeck, Germany), three of which were run in-house by the manufacturer (CBA-EI) and seven at other diagnostic centres (CBA-EO); four flow cytometry assays (FACS); three tissue-based assays using indirect immunofluorescence (2; TBA-IIF) or an optimised immunohistochemistry technique (1; TBA-IHC); and one commercial ELISA (Iason, Graz, Austria). All assay protocols (see online supplementary appendix I) and data from each test are provided (see online supplementary appendix II).
Supplementary appendix 1
Supplementary appendix 2
Assays
Statistics
The sensitivity, specificity, positive and negative likelihood ratios, and their 95% CIs were calculated using MedCalc V.15.8. Assay accuracy was calculated as (((true positives (TP)+true negatives (TN))/total samples)×100)). Intraclass correlation (ICC) of the semiquantitative score for all assays was used as a measure of agreement across the 21 assays using the Real Statistics Resource Pack software (Release 4.3; www.real-statistics.com) in patients with clinically definite NMO,16 excluding the minor criteria of AQP4-antibody seropositivity and on the whole cohort.
Results
Identification of specific assays using controls
Ninety-two coded control samples (37 MS, 39 headache and 16 other inflammatory disease) were sent randomly interspersed with the test samples. Overall, 16 of the 21 assays were >95% negative in the controls. Figure 2 displays the negative (white cells) and positive results (graded pink (low positive) to red cells (high positive)). Twelve assays were highly specific (0–1 false-positive test results; grouped on the left-hand side of figure 2) and included all three commercial CBAs performed in-house (CBA-EI, n=3), five of seven performed at different centres (CBA-EO, n=5), two of the three live CBAs, one of the four FACS assays and one of the three TBA. However, another nine assays produced from two to 10 positive results in this negative control cohort, with two assays (12 and 17) finding 9 and 10 positives. However, there was very little consistency in the results for any one serum between assays, supporting the ‘false positivity’ of the results, except for one sample from a patient with MG who was positive on six tests.

Assay specificity based on results from the 92 randomised control patient samples. These comprised 37 multiple sclerosis (35 relapsing remitting, 2 primary progressive), 1 connective tissue disease, 1 neuromyotonia, 1 progressive encephalomyelitis with rigidity and myoclonus, 4 clinically isolated syndrome, 1 acute disseminated encephalomyelitis, 1 Susac syndrome, 2 tumour (1 B-cell lymphoma, 1 colon carcinoma), 5 myasthenia gravis and 39 headache. Each column represents an individual assay (see table 2 for assay details) except for the first column which shows the serostatus assigned by the participating centre. Assays are grouped on the basis of their specificity in this cohort: assays on the left-hand side have 0 or 1 false-positive results (12 assays), whereas assays on the right-hand side have more than 1 false-positive result (9 assays). The assays are numbered 1–21: 1–3 are live cell-based assays (CBAs), whereas 4–6 are fixed CBA performed in-house at Euroimmun, 7–13 are fixed CBA performed at other European centres, 14–17 are flow cytometry assays, 18–20 are immunohistochemistry assays with detection based on enzymatic colour change (18) or fluorescence (19–20), and 21 is a commercially available ELISA (Iason). Each row represents a single serum sample. Positive results are coloured pink to red with a semiquantitative score from ‘1’ to ‘4’ inserted, whereas a negative result is white.
Defining AQP4-antibody-positive NMO
On the basis of the control data, any sample from patients with clinical NMO, ON or TM who were positive on two or more of these highly specific assays was considered seropositive NMO/SD. Results from the 50 patients whose clinical features fulfilled criteria for NMO independent of AQP4-antibody positivity are presented in figure 3. At least six of the highly specific assays (on the left-hand side of figure 3) identified 32/50 NMO sera (64%) as positive (all submitted as seropositive) and these were classified as AQP4-antibody-positive NMO, with the remaining 18 as AQP4-antibody seronegative NMO. All samples submitted as seronegative were negative on these specific assays. However, among the 18 seronegative samples, 10 were positive on at least one of the assays with lower specificity (right-hand side of figure 3), including one serum submitted as positive. Using the semiquantitative data provided by each centre (scoring samples from 0 (negative) to 4 (highly positive)), there was substantial concordance across all 21 assays for patients with clinically definite NMO (ICC of 0.753 (95% CI 0.669 to 0.831), with the complete data set giving an ICC of 0.820 (0.785 to 0.851).

Defining the serostatus of patients with neuromyelitis optica (NMO). Results in patients with NMO, defined by the 2006 Wingerchuk criteria excluding AQP4 serostatus. Results are presented as in figure 2, with each column representing an individual assay, apart from the first column that shows the serostatus submitted with the sample, and each row represents an individual serum. A positive result is graded in colour from pink to red with a semiquantitative score from ‘1’ to ‘4’ inserted. A negative result is displayed as a white box. Two individual results from patients with seropositive NMO were considered unevaluable by the testing centre and were scored negative. In total, 32 of 50 NMO samples are considered seropositive as they were positive on at least two of the specific assays. The remaining 18 samples were defined as seronegative NMO, including one that was submitted as seropositive. The numbers at the bottom of the figure show the assay sensitivity (%) based on these clinically defined patients.
Defining AQP4-antibody-positive NMOSD
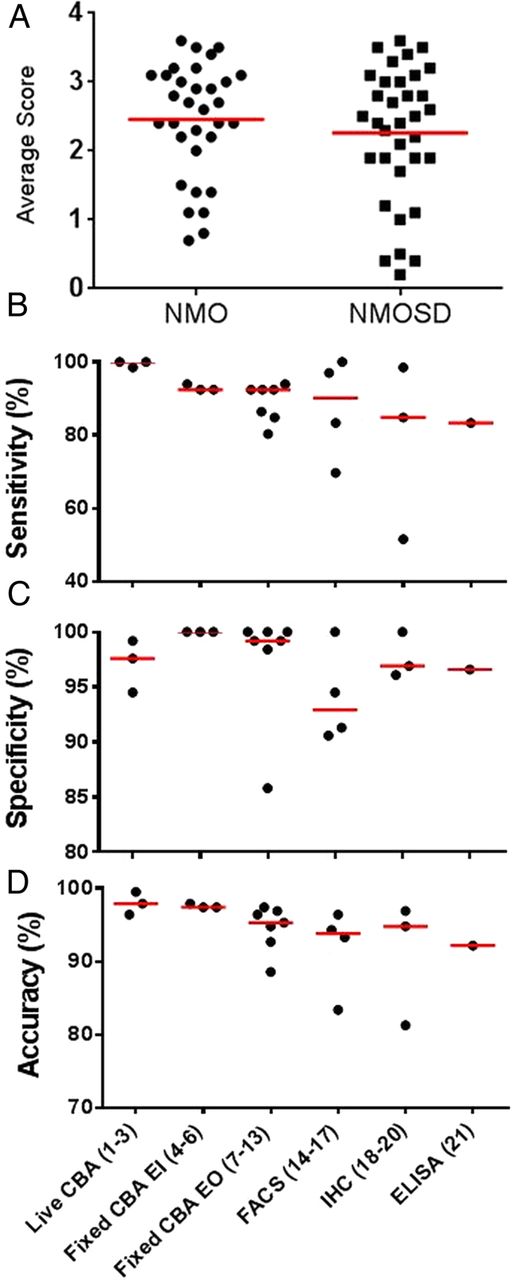
Very similar results were found in the 51 samples from patients with NMOSD (9 ON, 31 TM and 11 with ON and TM) who did not fulfil the criteria for NMO independent of antibody status. Thirty-one samples (61%) were positive on at least 4/12 assays that were highly specific (figure 4). However, there were six further sera that were identified as positive in only one or two specific assays and variably in the less-specific assays. Three were defined as AQP4-antibody positive as they were positive on two assays and the results were 100% concordant using a single technique (live CBA), while the remaining three samples that were positive on a single assay were considered negative. The NMOSD samples included nine from patients with NMO who required AQP4-antibody seropositivity to fulfil the 2006 Wingerchuk criteria, of which seven were positive in 18–21 of the 21 assays, and the other two were negative on all assays. Overall, the average antibody binding scores did not differ between the patients with NMO and NMOSD (figure 5A).

A heatmap of the entire data set presented in a similar fashion to figures 2 and 3. Each column is an individual assay. They are in the same order as in figures 2 and 3. Each row is an individual serum sample. Results are based on the semiquantitative scores; negative results are blue and positive results range from yellow (low positive) to red (high positive). The control samples are shown in (A) and the neuromyelitis optica (NMO) and NMO spectrum disorder (NMOSD) samples in (B). In total, 34 of 51 samples in the NMOSD are considered seropositive and 17 seronegative. The final serostatus of the NMO and NMOSD samples is listed on the right-hand side in B. The heatmap was generated using GENE-E V.3.0.204 (http://www.broadinstitute.org/cancer/software/GENE-E/).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Overall metrics of the AQP4 assays. Sixty-six samples were considered seropositive: 32 NMO and 34 NMOSD. Using the semiquantitative scores of 0–4 for each assay result, there was no difference in the average assay score across 21 assays between the NMO and NMOSD groups (A). (B–D) The assays are grouped by assay type on the x-axis with the study assay number in parentheses. The sensitivity (B) of assays was based on the samples classified as AQP4-antibody positive NMO or NMOSD (66 in total). The specificity (C) is based on the 92 control samples and the 35 seronegative NMO/SD samples. The accuracy (D) calculation was based on the categories described above: (((true positive+true negative)/total tests)×100). CBA, cell-based assay; IHC, immunohistochemistry; NMO, neuromyelitis optica; NMOSD, NMO spectrum disorder; SD, spectrum disorder.
Overall AQP4 assay metrics
Assay sensitivity was based on the 66 samples defined as seropositive (32 NMO, 34 NMOSD) and the specificity based on the 92 control samples and the 35 NMO (n=18) and NMOSD (n=17) samples defined as seronegative (table 3). The specificities, sensitivities and accuracy (((TP+TN)/total tests)×100) for the different types of assays are shown in figure 5B–D. Three assays were 100% sensitive: 2/3 live CBAs, and 1/4 FACS; all three were based on the transient expression of the human AQP4-M23 isoform. A further 10 assays were over 92% sensitive: the third live CBA (98.5%), a FACS assay (97.0%), a TBA that used optimised immunohistochemistry rather than immunofluorescence (TBA-IHC; 98.5%), and seven fixed CBAs (92.4–93.9%) and demonstrated excellent concordance. The remaining assays were between 51.5% and 86.4% sensitive including three fixed CBAs (80.3–86·4%), two FACS assay (69.7%, 83.3%), two TBA-IIF (51.5%, 84.8%) and the ELISA (83.3%); 4/6 of these assays that employ recombinant antigen used the AQP4-M1 isoform.
Assay metrics based on 66 AQP4 positive samples and 127 controls
Accuracy
If the sensitivity of the assays is based only on the results from the total NMOSD cohorts, without taking into consideration the results of the control samples, assays 12, 14 and 15 would be considered more sensitive (68–72%) than assays 1, 2 and 4 (62–64%) by an average of 6% (figure 2). If we include the specificity data from the control cohort, the accuracy of these two groups of assays is similar (77.8% vs 76.2%). However, this study design does not take into account ‘false-positive’ results within the correct clinical context found by assays 12, 14 and 15 (lower section of figure 2). The results from the seronegative NMO/SD samples were combined with the control sera results to measure assay specificity and assay accuracy was defined as (((TP+TN)/total tests)×100). In this instance, a TP result includes any positive result in the 66 patients with NMO/SD defined as seropositive and a TN result is any negative result in the control cohort or the seronegative NMO/SD patient samples (figure 5D). The difference in accuracy between these two groups is now clearer (99.3% vs 93.4%). Overall, the most accurate assays are the CBAs either on live cells or on the fixed CBA scored in-house by the company.
MG is a confounder for AQP4 assay studies
In this study, sera from five patients with MG without NMO/SD were included as negative control samples. However, a proportion of patients with MG go on to develop NMO. They can be seropositive for AQP4 antibodies many years before they develop clinical signs of NMO.21 Results in these patients are difficult to classify when evaluating AQP4 assay performance and should be omitted. To further illustrate this association, six of the AQP4 seropositive patients from four different centres had coexisting MG.
Discussion
Over the past 10 years, a number of new antibody-mediated CNS diseases have been discovered. The diseases are subacute and need to be diagnosed promptly in order to establish optimal treatments, but not all patients present with the full criteria for the clinical syndrome and may require antibody positivity for diagnosis; conversely, some patients fulfil clinical criteria but are persistently ‘seronegative’. Antibody assays, in sera or CSFs, have been developed in many laboratories, but there are no quality evaluation systems available yet, and both false-negative and false-positive results can mislead the clinician. Here, 15 European centres performed assays on the same randomised coded sera from 101 patients with NMO or NMOSD and 92 controls. Classification of the AQP4-antibody-positive cohort was based on patients with the correct clinical phenotype (NMO, ON, TM or ON+TM), and assays that were highly specific within the study. The selection process worked equally well on patients with NMO or NMOSD. The results support the high specificity and sensitivity of CBAs and of flow cytometry and immunohistochemistry in individual centres, and demonstrate the importance of multicentre studies to identify false-positive results within clinically defined cohorts.
We used NMO as the model disease because patients with NMO have a clinical phenotype that can be defined independent of antibody status and many laboratories are running the AQP4 antibody assays. However, an important feature of these assays is to identify accurately the patients with AQP4-antibody-negative NMO since they relapse less and may not require such aggressive immunotherapy.22–24 A study design using clinically defined cohorts to determine assay sensitivity does not identify the patients with AQP4-antibody-negative NMO and false-positive results in this cohort would lead to incorrect increases in assay sensitivity and specificity. This issue cannot be detected in single assay studies, but is highlighted when multiple assays are used on the same samples, and the conclusions based on coherence between results.
The random inclusion of samples from neurological patients who shared autoimmune or neurological features with NMOSDs was therefore an essential first step to identify the most specific assays. Having established the specificities with the disease control data, we could then identify the patients within the correct clinical context who were positive on at least two of the most specific assays and define them as seropositive. We were then able to define the patients with seronegative NMO, and use the same approach to define the patients with seropositive NMOSD, where there is the most clinical uncertainty. Addition of the patients with defined seronegative NMO and NMOSD to the control cohort improved the outcome measure of these assays for clinical use.
This post-study analysis also suggests a benefit of using the M23 isoform of AQP4 for optimal sensitivity with the top three ranked assays based on the transient expression of the human AQP4-M23 isoform. The live CBAs employ AQP4-M23 transfected cells and consistently perform well25 ,26 but are technically demanding and time-consuming, limiting their use except in specialist centres. Additionally, we do not have data from an AQP4-M1 live CBA within the study for direct comparison. The commercially available CBA is likely to become the standard for centres throughout the world; overall, it performed well in-house and in most other centres but issues at one or two centres need resolution. Perhaps surprisingly, the flow cytometry assays produced the greatest variations in sensitivity (69.7–100%) and specificity (90.6–100%); however, these can be explained by differences in assay methodology, sample processing and cut-off determination. The differences in data from centres using similar technologies suggest that experience with individual techniques impacts on the data produced. The flow method has the potential advantages of processing many samples together, establishing cut-offs based on multiple control sera and establishing independently any non-specificity of the sample (for instance, non-specific binding to the untransfected cells) all in a quantitative manner; hence, it should be pursued further. The immunohistochemistry on fixed tissue sections, originally used to define AQP4-IgG but considered poorly sensitive,26 was highly sensitive in one centre27 and could be used as an initial screening test with confirmation of positives by an antigen-specific CBA in centres where the costs of performing all assay requests on expensive commercial tests would be prohibitive.
This study design improves the assessment of AQP4 assays and provides a method to compare similar assays based on other targets. However, further study design improvements could be made. More control samples, particularly autoimmune samples, should be included but ambiguous patient cohorts, as is the case with patients with MG when studying AQP4 antibodies, should be considered separately. In one or two centres, individual assays (immunohistochemistry and flow cytometry) were excellent. In future studies, all assays to be tested should be implemented at multiple sites to evaluate assay reliability and reproducibility. In addition, multiple testing at a single centre may be helpful in adding confidence to test results in a routine clinical setting, but it could impact on interpretation of subjective results of the individual tests. Moreover, since all samples were sent in a single shipment, the assays had not necessarily been performed under conditions which would apply to the testing of samples that might be referred routinely. Continued assessment of a low number of samples sent intermittently to these laboratories through routine channels, as in external quality assurance services (EQAS), would be essential to monitor testing at all centres in the future.
The importance of antibody diagnostics is undoubted, with a large number of different antibodies now identified in a wide range of neurological diseases involving different forms of encephalitis or demyelinating/white matter conditions. For instance, identification of AQP4-antibodies is important to confirm a clinical diagnosis of NMO and ensure optimal treatment, as well as to define accurately those patients with partial phenotypes (NMOSDs) who are treated in a similar manner. In addition, greater emphasis is now placed on assay outcome in patient diagnosis according to the new NMOSD diagnostic criteria.28 False-positive results, highlighted in this study, need to be avoided because of the risk of overaggressive immunotherapies in patients with alternative diagnoses. These aspects will be even more important in diseases where there are no established clinical criteria such as in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies who can present with a phenotype similar to NMO but are less likely to suffer long-term disability,29 or anti-NMDA receptor encephalitis where the diagnosis partly depends on detection of the antibody.30 This study demonstrates the feasibility and advantages of performing multicentre comparisons of specificities and sensitivities and identifying and solving specific difficulties in the tests. The outcomes should improve the accuracy and confidence in AQP4-antibody testing, and suggest a way to carry out these studies in diseases where the clinical associations are less well established.
Acknowledgments
This study was carried out under the auspices of the Eugene Devic European Network (EDEN) project (ERA–Net ERARE 2: http://www.erare.eu/financed-projects/eden). All partners participated in this work in addition to 13 other invited centres involved in AQP4 antibody testing in Europe.
References
Footnotes
PW and MR are joint first authors
AV is a senior author
Contributors PW, MR, SS, AV and RM contributed to the concept or design of the manuscript. All authors were responsible for the acquisition, analysis or interpretation of the data. All authors contributed to revisions of the manuscript for important intellectual content and approved its final version.
Funding This study was funded by the Eugene Devic European Network (EDEN) project (ERA-Net ERARE 2: http://www.erare.eu/financed–projects/eden). Euroimmun AG provided assay kits for groups from Denmark and Hungary at 50% discount, and at no cost to both Italian groups. The work in Oxford was supported by the National Health Service National Specialised Commissioning Group for Neuromyelitis Optica and the National Institute for Health Research Oxford Biomedical Research Centre.
Competing interests AS is funded by Fundació la Marató de TV3 (101610). FT is enrolled in the doctoral programme SPIN funded by the Austrian Science Fund (FWF; project W1206). ZI is funded by Lundbeckfonden and Scleroseforeningen of Denmark. AB has received a grant from Bayer Healthcare on NMO. LZ would like to thank RH for her excellent technical assistance with the assay. RH was supported by a Schrödinger fellowship funded by the Austrian Science Fund (FWF; project J3230). CC acknowledges funding from Fundació la Marató de TV3 (493/C/2012). FP acknowledges funding from the Bundesministerium für Bildung und Forschung (Competence Network Multiple Sclerosis KKNMS). BW acknowledges funding from the Dietmar-Hopp-Stiftung and from Merck Serono. RM and AR acknowledge Nathalie Dufay from NeuroBioTec-Banques (Hospices Civils de Lyon, France) and funding from Association pour la Recherche sur la Sclerose en Plaques (ARSEP). PW has received speaker honoraria from Biogen Idec and Euroimmun AG; holds a patent with Oxford University for LGI1/CASPR2 antibodies, licensed to Euroimmun AG; and for GABAAR antibodies. MR reports other from University Hospital of Innsbruck (TILAK), during the conduct of the study; personal fees from Euroimmun, outside the submitted work. AS has received compensation for consulting services and speaking from Bayer–Schering, Merck–Serono, Biogen–Idec, Sanofi–Aventis, Teva Pharmaceutical Industries Ltd and Novartis. HHN has received travel funding and speaker honoraria from Novartis Healthcare, Biogen Idec, Genzyme Denmark and Teva Denmark. ZI reports grants from Lundbeckfonden, Denmark; Scleroseforeningen, Denmark, during the conduct of the study; personal fees from compensation for consulting services and speaking from Bayer–Schering, Biogen–Idec, Merck–Serono, Novartis, Sanofi–Aventis, and Teva Pharmaceutical Industries Ltd, outside the submitted work. TB reports non-financial support from Euroimmun AG, during the conduct of the study. KR is shareholder and an employee of Euroimmun AG. AB reports grants from Bayer Heathcare, personal fees from Biogen Idec, personal fees from Merck Serono, personal fees from Teva, personal fees from Novartis, personal fees from Genzyme, other from Biogen Idec, other from Novartis, other from Genzyme, other from Roche, other from Alexion Pharmaceuticals, outside the submitted work. LG received congress and travel compensations from Euroimmun. AB received honoraria for serving in the scientific advisory boards of Almirall, Bayer, Biogen Idec, Genzyme, and received speaker honoraria from Biogen Idec, Genzyme, Novartis, TEVA with approval by the Director of AOU San Luigi University Hospital; his institution has received grant support from Bayer, BiogenIdec, Merck, Novartis, TEVA from the Italian Multiple Sclerosis Society, Associazione Ricerca Biomedica ONLUS and San Luigi ONLUS. LZ reports personal fees from Teva Pharmaceutical, personal fees from Novartis, outside the submitted work. MC has received compensation for consulting services and speaking honoraria from Bayer Schering Pharma, Merk Serono, Biogen-Idec, Teva Pharmaceuticals, Sanofi-Aventis, Genzyme, and Novartis. XM has received speaking honoraria and travel expenses for scientific meetings, has been a steering committee member of clinical trials or participated in advisory boards of clinical trials in the past years with Bayer Schering Pharma, Biogen Idec, EMD Merck Serono, Genentech, Genzyme, Novartis, Sanofi-Aventis, Teva Phramaceuticals, Almirall and Roche. MT has received compensation for consulting services and speaking from Bayer-Schering, Merk-Serono, Biogen-Idec, Teva, Sanofi-Aventis, and Novartis. AS reports receiving grants from the Scientific and Technological Research Council of Turkey—Health Sciences Research (grants numbers 109S070 and 112S052); and also unrestricted grants from Bayer-Schering AG and Merck-Serono to their Neurology Department Clinical Neuroimmunology Unit. AS also reports receiving honoraria or consultation fees Biogen Idec, Novartis and Teva. He had received travel and registration coverage for attending several national or international congresses or symposia, from Merck Serono, Biogen Idec, Novartis, Teva, Bayer-Schering and Allergan. AA received grants to her department from The Scientific and Technological Research Institute of Turkey-Health Sciences (grants numbers 109S070 and 112S052), and she also received honoraria, research and travel grants from Teva, Merck-Serono, Gen Ilac, Bayer-Schering, Novartis. FP has received travel funding, speaker honoraria and personal compensation for activities on advisory boards and steering committees from Bayer, Biogen Idec, Alexion, Chugai, MedImmune, MerckSerono, Novartis, Teva, and Sanofi Genzyme. He has received grants from the Guthy Jackson Charitable Foundation and the National Multiple Sclerosis Society of the USA. OA reports grants from German Ministry for Education and Research (BMBF), during the conduct of the study; German Research Foundation (DFG); Hertie Foundation; Biogen; Novartis; Bayer; outside the submitted work; and Honoraria for consultancy and speaking by Biogen, Novartis, Bayer Schering, Teva, Chugai, Genzyme, MedImmune, and Merck Serono. BW reports personal fees from Bayer Healthcare, grants and personal fees from Biogen, Merck Serono; Genzyme, a Sanofi Company; Novartis Pharmaceuticals; TEVA Pharma; grants from Biotest, German Ministry of Education and Research; Dietmar Hopp Foundation; outside the submitted work. H-PH reports personal fees from MedImmune, outside the submitted work. MP is an employee of and holds stock in Euroimmun AG. PT reports non-financial support from Roche Pharmaceuticals, outside the submitted work. SS is an employee of Euroimmun AG. AV is on the Advisory Board for Neurology; was an associate editor for Brain; holds patents with Oxford University for MuSK, LGI1/CASPR2, and GABAAR antibodies, and receives a proportion of royalties. RM reports personal fees from MedImmune, personal fees and non-financial support from Biogen, non-financial support from Merck-Serono, non-financial support from Teva, non-financial support from Novartis, non-financial support from Sanofi, outside the submitted work.
Ethics approval The research use of referred sera was approved by the Oxfordshire Research Ethics Committee (reference number 10/H0606/56), by the Ethical Review Board of the University of Heidelberg, Germany, by the Regional and National Ethical Committee of Hungary (3893.316–12464/KK4/2010 and 42341–2/2013/EKU, Hungary), by the Ethics Committee of the Region of Southern Denmark (ref S–20120066), by the French data protection authority, by the regional committee for medical and health research ethics, Western Norway (REK#3.2006.1235), following IRB approval in Berlin, Dusseldorf and Munich, Germany, and according to the Dutch regulation for use of patient material.
Provenance and peer review Not commissioned; externally peer reviewed.