Article Text

Abstract
Background Parkinson’s Disease (PD) psychosis refers to the spectrum of illusions, formed hallucinations and delusions that occur in PD. Visual hallucinations and illusions are thought to be caused by specific cognitive and higher visual function deficits and patients who develop such symptoms early in the disease course have greater rates of cognitive decline and progression to dementia. To date, no studies have investigated whether such deficits are found prior to the onset of PD psychosis.
Method Here we compare baseline cognitive, biomarker (structural imaging and cerebrospinal fluid) and other PD psychosis risk factor data in patients who go on to develop illusions or hallucinations within 3–4 years of follow-up in the Parkinson’s Progression Markers Initiative cohort of newly diagnosed PD.
Results Of n=423 patients with PD, n=115 (27%) reported predominantly illusions with the median time of onset at 19.5 months follow-up. At study entry these patients had reduced CSF amyloid Aß1-42, lower olfaction scores, higher depression scores and increased REM sleep behaviour disorder symptoms compared to patients without early onset PD psychosis but no differences in cognitive, higher visual or structural imaging measures. A subset of patients with early onset formed hallucinations (n=21) had reduced higher visual function at baseline, cortical thinning in parietal, occipital and frontal cortex and reduced hippocampal volume.
Conclusions The findings suggest early onset illusions and formed hallucinations are linked to amyloid pathology in PD and point to a difference in the underlying pathophysiological mechanism of illusions and formed hallucinations, with implications for their respective links to future cognitive decline.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Parkinson's disease (PD) psychosis refers to a spectrum of symptoms that occur at different disease stages.1 ,2 Minor symptoms occurring early in PD include visual illusions (a real object perceived incorrectly), passage hallucinations (a hallucinated animal or figure passing in the peripheral visual field) and presence hallucinations (the sense of someone nearby). In some patients, such experiences precede the onset of motor symptoms.3 As PD progresses, formed visual hallucinations occur with preserved insight (recognition that the experiences are not real). In later PD stages, insight may become lost with the development of false explanations about the experiences (secondary delusions), hallucinations in other sensory modalities (auditory, tactile or olfactory), delusions (eg, themes of guilt or grandiosity4) and misidentification syndromes (eg, the belief that someone familiar has been replaced by an imposter5). Such symptoms are an important treatment priority as they precipitate the move into a care home from living independently6 ,7 and impact on quality of life.8
Alongside clinical considerations, PD psychosis is emerging as an important biomarker of disease progression and cognitive outcome. Although most PD patients will develop hallucinations as disease progresses, an estimated 60% experiencing PD psychosis by 12 years,9 a distinction can be made between patients who develop such symptoms early in the disease course and those who develop it in later stages. Several studies have found visual hallucinations predict later dementia or greater cognitive decline.10–13 The underlying mechanism of this association is unclear, but cross-sectional studies have identified higher visual function deficits in domains such as object and visuospatial perception and cognitive deficits in domains of attention, memory and executive function in PD patients with visual hallucinations compared with those without them.14 These deficits of visual perception and attentional control are thought to cause visual hallucinations and illusions through an imbalance of top-down expectations and bottom-up visual processing (see ref. 15 for a review). Consistent with the neuropsychological findings, visual hallucinations are associated with reduced grey matter volume in the visual system, hippocampus, frontal regions (particularly lateral frontal cortex) and cerebellum.16–20 Lewy bodies, amyloid and tau pathologies are found at postmortem in some of these regions in PD patients with a history of visual hallucinations,21 although such pathology is notably absent in the cortex and hippocampus of non-demented PD patients with visual hallucinations.22
While the link between formed hallucinations and visual and cognitive deficits is clear, what is less certain is whether the same is true of illusions, the earliest symptoms of PD psychosis. Patients with illusions, presence and passage hallucinations do not have significant differences in verbal memory, verbal fluency or sustained attention compared with patients without such symptoms23 and the only regions of cortical volume loss identified in an imaging study of early symptoms were in the midbrain, cerebellar vermis and visual parietal cortex.24 Furthermore, other risk factors for PD psychosis have been identified whose role is unclear, including rapid eye movement (REM) sleep behaviour disorder (see ref. 25 for a review), depression26 and dopamine agonist medication.9 ,13 Such findings raise the possibility that the earliest symptoms of PD psychosis may differ in their aetiological cause to later symptoms. Here, we explore this issue and the wider implications of early-onset and late-onset PD psychosis by examining the profile of cognitive, biomarker (structural imaging and cerebrospinal fluid (CSF)) and other risk factors prior to the onset of illusions and hallucinations in the Parkinson's Progression Markers Initiative (PPMI) data set.27
Methods
The Parkinson's Progression Markers Initiative (PPMI)27 is an observational multicentre study of newly diagnosed, drug-naïve patients and healthy controls using standardised, clinical, imaging, CSF and cognitive assessments at 3-month intervals in the first year and 6-month intervals in subsequent years (http://www.ppmi-info.org/). The analysis reported here was performed on the PPMI data set completed up to 15 January 2016 containing n=195 healthy controls and n=423 PD patients based on clinical and DAT scan criteria (patients classified as having scans without evidence for dopaminergic deficit are not included in the analysis). At the time of download, 70% of PD patients and 100% of healthy controls had been followed for 3 years or more. Subsets of the PD and healthy control groups were created for the analysis as described below.
Ethics Each participating PPMI site received approval from an ethical standards committee on human experimentation before study initiation and written informed consent for research was obtained from all participants in the study.
Group definitions
PD patients were divided into groups based on the subsequent development of PD psychosis during PPMI follow-up using the UPDRS part 1 hallucinations/psychosis item. The item categorises symptoms experienced over the preceding week into 5 levels: 0, no symptoms; 1, minor illusions and non-formed hallucinations; 2, formed hallucinations with preserved insight; 3, formed hallucinations without insight; 4, psychosis with delusions and hallucinations. Four groups were defined:
HC Healthy controls.
PD-Psy0 PD patients without reports of psychosis symptoms at any assessment. An arbitrary minimum period of follow-up (18 months) was used as an additional inclusion criterion to exclude patients for whom the presence or absence of early-onset PD psychosis symptoms was, as yet, unknown.
PD-Psy1+ PD patients reporting any PD psychosis symptom graded 1 or more on the UPDRS hallucinations/psychosis item at one or more assessments.
PD-Psy2+ A subgroup of patients in the PD-Psy1+ group reporting symptoms graded 2 or more on the UPDRS hallucinations/psychosis item at one or more assessments.
Assessments
Cognitive function measures The Montreal Cognitive Assessment (MoCA) assessed general cognitive abilities (maximum value=30). Mild cognitive impairment in PD (PD-MCI) was assessed using level 1 criteria of MoCA score <26.28 Executive function was assessed using semantic fluency (the sum of item names per minute in the animal, vegetable and fruit categories). Verbal memory was assessed using the Hopkins Verbal Learning Test delayed recall score (20 min delay; maximum score=12). Visuospatial function was assessed using the Benton Judgement of Line Orientation (maximum value=15). Attention was assessed using Letter Number Sequencing (maximum=21).
Motor assessments
Motor symptoms were assessed using the UPDRS part 3 motor subscale.
Other risk factors REM Sleep Behaviour Disorder Screening Questionnaire (maximum=12). Geriatric Depression Scale (maximum=15; higher score lower mood). As decreased olfaction score has been associated with cognitive decline in the PPMI data set,29 the University of Pennsylvania Smell Identification Test was also included in the analysis (UPSIT; maximum score 40).
Structural imaging T1-weighted images were acquired on a 3 T Siemens scanner using a magnetisation-prepared rapid-acquisition gradient echo sequence. For details on the PPMI MRI protocol, see Pereira et al.30 A subset of PD patients and healthy controls had baseline MRI data of sufficient quality to estimate cortical thickness (HC=48; PD-Psy0=87; PD-Psy1+=31; PD-Psy2+=9). As the number of patients with usable baseline scans in each group was small, to maximise numbers and differences between groups, the PD-Psy2+ subset of patients was not included in the PD-Psy1+group and 6 patients in the PD-Psy0 group that had withdrawn before 18 months follow-up were included in the analysis. Images were preprocessed and analysed using Freesurfer (V.5.3, http://freesurfer.net/) to: (1) generate a cortical surface model estimating cortical thickness at each vertex, smoothed with a 15 mm FWHM kernel and (2) estimate the volume of subcortical grey matter structures.
CSF methods Details of the PPMI CSF sampling and analysis procedures are described on the study website (http://www.ppmi-info.org/). CSF Aβ1-42, total tau, p-tau1818 and α-synuclein values were used in the analysis. As α-synuclein is present in red blood cells, CSF haemoglobin was examined as a measure of CSF blood contamination.
Statistical analysis
Baseline demographic, PD motor, cognitive and non-cognitive data were compared using t-tests, non-parametric or χ2 tests as appropriate within SPSS V.20. Differences in cortical thickness between groups were assessed using Qdec within FreeSurfer with Monte Carlo simulations (p<0.05), while controlling for the effects of age, gender, education, presence of PD-MCI (see ref. 30 for criteria), UPDRS motor scores (when appropriate), differences in scanner site and software. Differences in subcortical grey matter volumes were evaluated using an analysis of covariance in SPSS, while controlling for the covariates above in addition to intracranial volume as estimated by FreeSurfer.
Results
At the time of data download, n=115 (27%) PD patients had reported psychosis symptoms in one or more of the PPMI follow-up assessments over 4 years (PD-Psy1+ group). By comparison, only n=3 (1%) patients in the Healthy Control group reported any psychosis spectrum symptom. A subset of PD psychosis patients in whom no PD psychosis symptoms had been reported in any follow-up assessment was identified (PD-Psy0, n=286; 72% of the group had 3 years or more without PD psychosis symptoms) as well as a subset of healthy controls with 3 years follow-up (HC, n=144).
PD psychosis symptom onset and course
Figure 1 shows the number of patients in the PD-Psy1+ group over the 4-year follow-up period. The median time to the first PD psychosis symptom was 19.5 months. The majority of patients in the PD-Psy1+ group (81%) reported only illusions at any assessment (all UPDRS hallucination/psychosis scores=1). Fifty-eight per cent of PD-Psy1+ had two or more assessments with PD psychosis symptoms. A subgroup of PD-Psy1+ patients (PD-Psy2+, n=21) reported formed hallucinations or delusions at one or more assessments (one or more UPDRS hallucination/psychosis scores=2, 3 or 4). Of these patients, n=16 reported illusions on some assessments and formed hallucinations on others (illusions preceded formed hallucinations in n=9).
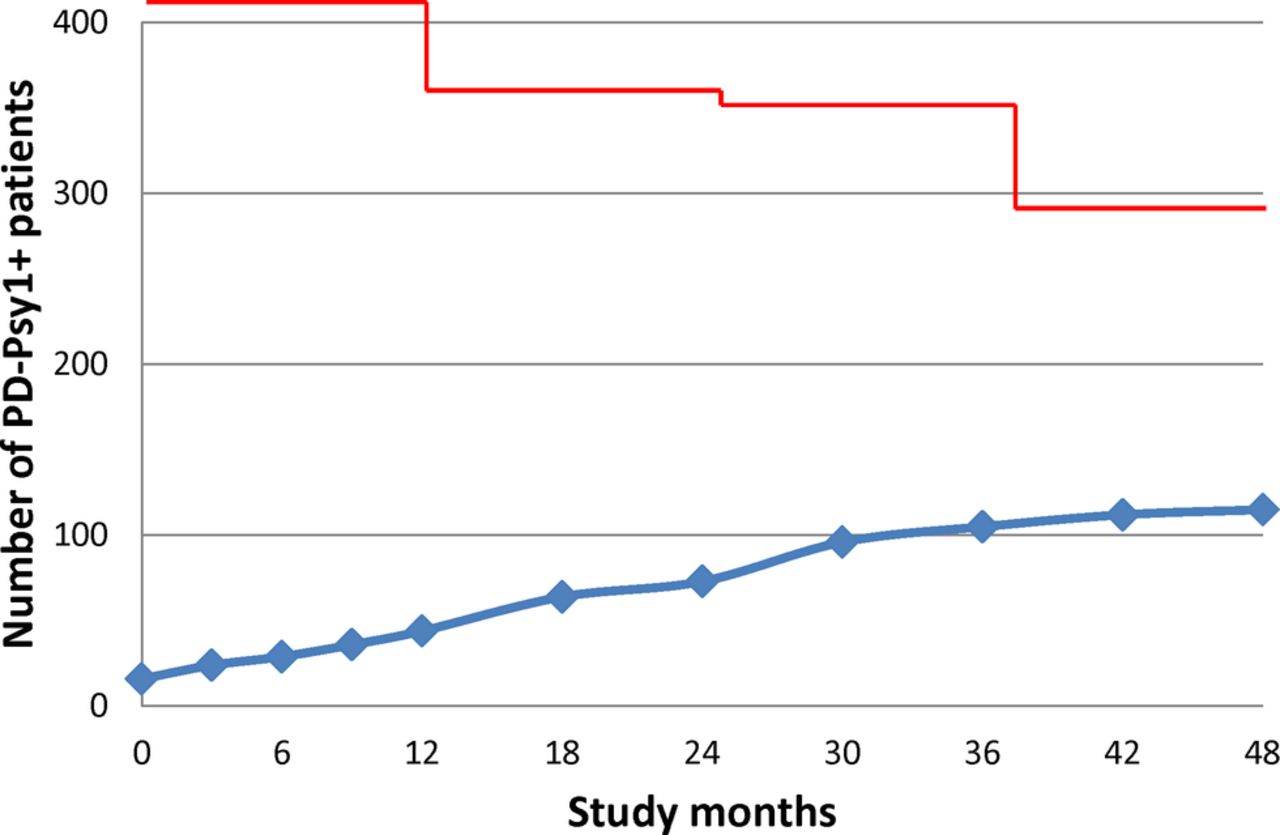
Number of Parkinson's disease (PD) patients in the PD Psy1+ group at each follow-up assessment. Y-axis scaled to total number of patients with PD at study entry (n=423). The red line indicates the number of patients that had completed baseline, 1 year, 2 year and 3 year assessments at the time of data download. The percentage of patients with PD psychosis symptoms may increase as further follow-up data is collected.
Table 1 shows demographic and risk factor characteristics at study entry for each group. There are no differences between PD patients without (PD-Psy0) and with early-onset psychosis spectrum symptoms (PD-Psy1+ or PD-Psy2+) on demographic or PD motor measures. RBD score, depression score and olfaction score differed between patients with and without early-onset PD psychosis (all p<0.05). There were no differences in cognitive measures between PD-Psy0 and PD-Psy1+ or the proportion of patients meeting level-1 criteria for PD-MCI.28 PD-Psy2+ had significantly reduced performance in the Benton Judgement of Line Orientation task compared with PD-Psy0. Consistent with previous studies, verbal fluency was reduced in PD-Psy2+ compared with PD-Psy0, although only at trend significance (p=0.07). We also looked for differences in cognitive scores in the period immediately before the onset of PD psychosis. Figure 2 shows the time course of MoCA score prior to PD psychosis onset (black circle) in patients with symptom onset at 1 year (green line), 2 years (red line) and 3 years (blue line) after study entry and the PD-Psy0 group (black line). Using a mixed model ANOVA with fixed factors TIME (months prior to hallucination onset in PD-Psy1+ or months after study entry in PD-Psy0) and GROUP (PD-PSy0, PD-Psy1+), we found no significant effect of GROUP (p>0.05) or a TIME by GROUP interaction (p>0.05) suggesting MoCA score or MoCA score decline prior to the onset of PD psychosis were equivalent in the PD-Psy0 and PD-Psy1+ groups. There were no TIME by GROUP interactions for fluency, Hopkins Verbal Learning, Benton Judgement of Line Orientation or Letter Number Sequencing (all p>0.05).
Baseline demographic Parkinson's disease motor, cognitive and other measures in each group

Global cognition and the onset of Parkinson's disease (PD) psychosis. The MoCA score trajectory prior to onset of PD psychosis (black circle) in subgroups of patients with PD psychosis onset at 1 year (green line), 2 years (red line) and 3 years (blue line). The trajectory of patients without PD psychosis (PD-Psy0) is shown in black. The slopes of MoCA decline do not differ between groups with and without PD psychosis (see text for further details).
PD psychosis and medication for PD
Patients at PPMI study entry are not taking medication which is only added when clinically appropriate. If the onset of PD psychosis is linked to PD medication, one might expect a higher proportion of patients with PD psychosis to be taking medication at any given time. As shown in table 2, a similar proportion of patients with and without early-onset PD psychosis are prescribed any medication for PD, L-Dopa or a Dopamine agonist in each follow-up period. Across all time points, the only significant difference was found at 24 months where a higher proportion of PD-Psy1+ was prescribed dopamine agonists. There is no difference between PD-Psy0 and PD-Psy1+ groups in the time from PD diagnosis to starting any PD medication or to starting a dopamine agonist (both p>0.05) (table 3). Surgical treatment for PD using deep brain stimulation (DBS) had been started in seven patients during follow-up (36–54 months after study entry; n=4 in PD-Psy0; n=3 in PD-Psy1+). Illusions preceded DBS in all three PD-Psy1+ patients by a minimum of 2 years. There is currently insufficient post-DBS follow-up evidence to comment on whether DBS improves or exacerbates PD psychosis symptoms.
Parkinson's disease (PD)-psychosis and medication for PD
Parkinson's disease-psychosis and time to medication onset
Structural imaging
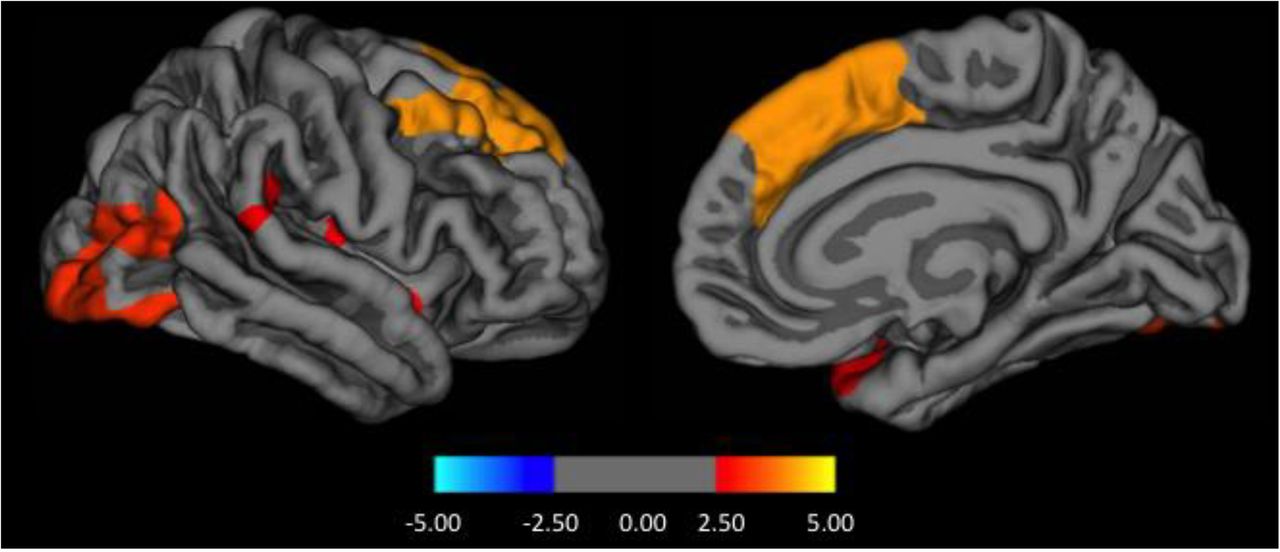
No significant differences in baseline cortical thickness were found between PD-Psy0 and PD-Psy1+ or between healthy controls and PD-Psy1+. However, compared with PD-Psy0, the PD-Psy2+ group had reduced cortical thickness in the right hemispheric frontal, occipital, parietotemporal (supramarginal gyrus) and insula regions and reduced volumes bilaterally in the basal ganglia and hippocampus (figure 3 and table 4).
Regions of cortical and subcortical thinning in patients with formed hallucinations

{kind=link}
{kind=link}
{kind=link}
Baseline cortical thickness differences between patients with formed hallucinations (PD-Psy2+) compared with those without Parkinson's disease (PD) psychosis (PD-Psy0) on a surface rendered right hemisphere in lateral (left panel) and medial (right panel) view. Colour coding indicates cluster significance on a logarithmic scale of p values (−log10).
Cerebrospinal fluid
Patients with early-onset PD psychosis (PD-Psy1+ and PD-Psy2+ groups) had decreased baseline CSF Aβ1-42 compared with patients without PD psychosis (PD-PSy0) (see table 5; MoCA score is equivalent across groups and has therefore not been included as a covariate in the analysis). There were no group differences in the levels of total tau, p-tau181 and α-synuclein. Red blood cell contamination as measured by haemoglobin concentration was evident but equivalent in patients with and without PD psychosis (p>0.05; Mann-Whitney U test).
Baseline cerebrospinal fluid measures in each group
Discussion
This is one of the first studies exploring clinical and imaging risk factors for early psychosis in a large cohort of de novo PD patients. In the PPMI data set, first-onset PD psychosis symptoms were reported at some point during 4 years of follow-up in 27% of PD patients. While the clinical and cognitive outcomes of these patients will be assessed at study completion, questions related to the onset of PD psychosis symptoms can be addressed in data already available. In what follows, we discuss our findings in relation to the aetiological mechanism of early-onset PD psychosis and its prognostic implications.
CSF Aβ1-42 and PD psychosis
We found an association between early-onset PD psychosis and reduced Aβ1-42,, but not with total tau, p-tau or α-synuclein. PD psychosis in later disease stages has been linked to higher levels of amyloid, tau and α-synuclein pathology in frontal, parietal and hippocampal areas21 and our finding may suggest early-onset PD psychosis is a biomarker for the subsequent development of Alzheimer pathology. Previous studies of the PPMI cohort have found reduced Aβ1-42 (but not total tau) associated with PD-MCI and reduced α-synuclein associated with executive dysfunction.31 A link between Aβ1-42, PD-MCI and early-onset PD psychosis might explain the poor longer term cognitive outcomes found in previous studies of visual hallucinations, although the relationship with PD-MCI is complex as the proportion of patients with PD-MCI did not differ between groups with and without PD psychosis in our data. Lower olfaction scores have also been associated with lower Aβ1-42 in the PPMI data set29 and our findings suggest the olfaction changes are also associated with subsequent development of PD psychosis. The neurobiological significance of this association is unclear.
Cognition, cortical changes and visual hallucinations
Cross-sectional studies of patients susceptible to visual hallucinations typically find deficits of executive function, attention, memory and visual perception (see ref. 14 for a review). We only found evidence for deficits in visuospatial processing (assessed by the Benton Line Judgement test) and trend level deficits in executive function. The lack of prominent deficits in other cognitive domains may reflect the small number of patients with formed hallucinations and differences in tests used in PPMI compared with previous studies of visual hallucinations.
The deficits of visual and cognitive function in PD patients with visual hallucinations are thought to relate to volume loss in frontal, parietal, limbic, occipital and hippocampal cortex (see refs 32–34 for recent reviews). Here, we found evidence for these structural changes in patients with de novo PD who go on to develop formed hallucinations. A notable difference from previous studies was the lack of prominent involvement of dorsolateral prefrontal cortex, inferior frontal cortex and ventral occipitotemporal cortex. The underlying pathology of these cortical and hippocampal changes is unclear as there is only sparse cortical Lewy pathology in patients with preserved cognition22 equivalent to that in the PPMI cohort. The overall distribution of cortical thinning found in patients with formed hallucinations also differs from that associated with PD-MCI in the PPMI cohort.30 Together with the control for PD-MCI status in our MRI analysis, this suggests the distribution of cortical thinning identified here may be specific to formed hallucinations.
Illusions and hallucinations: one aetiological mechanism or two?
Given that illusions and formed hallucinations are thought to share the same mechanism of dysfunctional top-down and bottom-up integration,35 an unexpected finding was the lack of cognitive differences or cortical thinning in the broadly defined PD psychosis group with predominantly illusions. This does not seem to reflect cognitive decline between the time of assessment and the time of symptom onset as there was no difference in the trajectory of MoCA global cognitive decline between groups before symptoms began. Although subtle cognitive and visual perceptual changes at baseline or symptom onset cannot be ruled out, the lack of difference between groups may suggest illusions and other early phenomena in PD have an underlying aetiological mechanism that differs from formed hallucinations. In support of this view, previous studies have found that presence hallucinations are associated with sleep regulation and somnolence scores while illusions are associated with somnolence and motor severity scores.36 These early symptoms have been linked to brainstem, eye movement and motion perception mechanisms in the visual parietal lobe24 ,37 rather than the ventral occipitotemporal cortical regions thought to underlie formed visual hallucinations.38 Our finding of increased REM sleep behaviour disorder score in the group with predominantly illusion symptoms is consistent with this view.
Other risk factors
We found a difference in the proportion of patients prescribed dopamine agonists at 2 years consistent with previous findings from the PPMI cohort.39 However, drug-naïve patients with PD psychosis in the PPMI cohort and other studies3 suggest that the onset of PD psychosis is not directly related to starting PD medication. The neurobiological interpretation of the association between higher baseline depression scores and early-onset PD psychosis is also unclear. An association between depression and illusions, passage and presence hallucinations in PD has been described previously;40 however, the evidence presented here suggests that depressive symptoms are not merely a response to illusions as, in the majority of patients, depression scores are elevated before illusion onset.
The timing of early-onset versus late-onset PD psychosis
As the prevalence of PD psychosis increases with disease duration, a high proportion of PD patients will eventually develop such symptoms. The distinction made in this study is between PD patients with onset of symptoms within the first 3–4 years of PPMI follow-up and ‘late onset’ patients in whom PD psychosis symptoms have not yet developed. The majority of the late-onset group have remained without PD psychosis symptoms for 3 years or more at the time of data download. Whether (or when) this group will develop psychosis symptoms is currently unclear but will help define the time window for late-onset PD psychosis.
Limitations
An important limitation to the study is the lack of phenomenological detail with respect to PD psychosis. For example, it is unclear whether formed hallucinations are visual or involve other sensory modalities or what symptoms are classed as illusions. However, previous studies41 allow us to predict with confidence that the symptoms were visual. Another limitation is the fact that information is only collected with respect to symptoms in the week prior to a given assessment. Symptoms occurring between assessments are thus missed in each group (although a strength of the PPMI study is the short interval between assessments in the first year), so that prevalence estimates of PD psychosis may be higher than those reported. The group distinctions might therefore be better characterised as low (PD-Psy0) and high (PD-Psy1+ and PD-Psy2+) rates of early-onset PD psychosis rather than groups without and with early-onset PD psychosis.
Conclusions
PD patients who develop illusions or hallucinations within 3–4 years of PD diagnosis have CSF amyloid reduction, olfactory impairment and increased depression and sleep behaviour scores that predate PD psychosis onset. The neurobiological implications of these factors and how they relate to each other and to prospective cognitive decline remain unclear. The findings also suggest illusions and formed hallucinations indicate different profiles of visual, cognitive, cortical and hippocampal involvement in PD. Patients who went on to develop formed hallucinations had visual function deficits, trend-level executive function deficits, widespread cortical thinning and hippocampal volume loss at baseline compared with patients without early-onset symptoms, but no differences were found in patients who went on to develop only illusions. If formed hallucinations imply the presence of neuropsychological deficits, cortical thinning and hippocampal volume loss, their prognostic implication for cognitive decline may differ from that of illusions. These differing biomarker implications highlight the need for scales and instruments that distinguish between illusions, formed hallucinations and other early symptoms in prospective studies of cognitive outcome.
Acknowledgments
Data used in the preparation of this article were obtained from the Parkinson's Progression Markers Initiative database (http://www.ppmi-info.org/). The authors thank Dr Fabrizia D'Antonio for help with R programming, the NIHR Maudsley Biomedical Research Centre and Dementia Unit and NIHR Programme Grants for Applied Research (RP-PG-0610-10100 SHAPED) for supporting involvement in this work. Dag Aarsland is a Royal Society Wolfson Research Merit Award Holder and thanks the Wolfson Foundation and the Royal Society for their support.
References
Footnotes
Disclaimer The views expressed are those of the authors and not necessarily those of the NHS, NIHR or the Department of Health.
Contributors All authors contributed to the planning and design of the study and approved the final manuscript. DHF and DA analysed the data and wrote the initial draft. JBP analysed the MRI data.
Competing interests None declared.
Ethics approval Each participating PPMI site received approval from an ethical standards committee on human experimentation before study initiation and written informed consent for research was obtained from all participants in the study.
Provenance and peer review Not commissioned; externally peer reviewed.