Article Text

Abstract
Lower motor neuron (LMN) syndromes typically present with muscle wasting and weakness and may arise from pathology affecting the distal motor nerve up to the level of the anterior horn cell. A variety of hereditary causes are recognised, including spinal muscular atrophy, distal hereditary motor neuropathy and LMN variants of familial motor neuron disease. Recent genetic advances have resulted in the identification of a variety of disease-causing mutations. Immune-mediated disorders, including multifocal motor neuropathy and variants of chronic inflammatory demyelinating polyneuropathy, account for a proportion of LMN presentations and are important to recognise, as effective treatments are available. The present review will outline the spectrum of LMN syndromes that may develop in adulthood and provide a framework for the clinician assessing a patient presenting with predominantly LMN features.
- NEUROPATHY
- NEUROPHYSIOLOGY
- NEUROIMMUNOLOGY
- GENETICS
- MOTOR NEURON DISEASE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Lower motor neuron (LMN) syndromes are clinically characterised by muscle atrophy, weakness and hyporeflexia without sensory involvement. They may arise from disease processes affecting the anterior horn cell or the motor axon and/or its surrounding myelin. Neuromuscular junction pathology and muscle disorders may mimic a LMN disorder and form part of the differential diagnosis.
LMN syndromes can be broadly classified as hereditary, sporadic or immune-mediated. Immune-mediated neuropathies, such as multifocal motor neuropathy (MMN) and chronic inflammatory demyelinating polyneuropathy (CIDP) are important to distinguish from sporadic and hereditary forms, as treatments are available. LMN presentations of motor neuron disease (MND) are most often sporadic, but several genetic mutations have been described which can be associated with LMN preponderance. Other hereditary forms of LMN syndromes include the spinal muscular atrophies (SMAs) and distal hereditary motor neuropathies (dHMNs). The increasing availability of next-generation sequencing (NGS), including the ability for multiple genes to be sequenced in parallel, has resulted in an increase in the discovery of novel genetic mutations.
The clinical evaluation of a patient presenting with a LMN syndrome includes a thorough assessment of disease onset and progression. This is particularly important to ascertain as a rapid rate of decline may support a diagnosis of MND and remains an important factor in distinguishing MND from other relatively indolent conditions, such as SMA and immune neuropathies. The pattern of weakness should be documented, including (1) symmetry versus asymmetry, (2) proximal versus distal involvement, (3) upper versus lower limb predominance and (4) presence versus absence of bulbar involvement. Nerve conduction studies and electromyography (EMG) are essential to confirm that the disorder is neurogenic and should focus on assessing (1) the pattern of involvement, including symmetry and length dependence, (2) presence of focal motor conduction block or demyelinating features and (3) the presence or absence of subclinical sensory abnormalities. Imaging, genetic testing, antibody markers and advanced neurophysiological techniques are useful adjuncts and form an extension of the clinical assessment. The present review will examine LMN syndromes from a clinical perspective as well as providing an overview of current understanding of pathophysiological mechanisms.
Late-onset SMA
SMA represents a group of genetic disorders resulting in the degeneration of anterior horn cells in the spinal cord and motor nuclei in the brainstem causing progressive, predominantly proximal muscle weakness with reduced or absent reflexes. They are classified into four types on the basis of age of onset and clinical course (SMA I–IV).1 SMA I and II are defined by onset in infancy. SMA III is a milder phenotype with signs of weakness presenting at or after 1 year of age with patients attaining the ability to walk unaided.1 It is associated with significant variability in the age of onset, disease progression and ambulatory period with some patients only developing walking difficulties in adulthood.2 Adult-onset SMA (SMA IV) typically presents in the third or fourth decade of life with a slowly progressive and relatively benign course.3 Respiratory insufficiency may occur in SMA IV, but is usually mild and life expectancy is normal.1
The vast majority of SMA is autosomal recessive (AR) in inheritance and related to mutations in the SMN1 gene located on chromosome 5q13. Most cases are homozygous for a deletion of exon 7 (94%), but a small percentage are compound heterozygous for a deletion in SMN1 and an intragenic mutation of SMN1.4 Targeted molecular genetic testing is the first-line investigation for SMA to detect homozygous deletions of SMN1 exon 7 gene. However, if only a single deletion is detected, sequencing the SMN1 gene should be performed to assess for a point mutation. Overall, 4–5% of patients with clinically typical SMA have no identifiable mutation in SMN1.5 Non-5q SMA can be inherited in an autosomal dominant (AD), AR or X-linked pattern with marked clinical and genetic heterogeneity. NGS technology has facilitated the discovery of a number of non-5q causative genes associated with SMA.6
Spinobulbar muscular atrophy (Kennedy's disease)
Spinobulbar muscular atrophy or Kennedy's disease is the most common adult-onset SMA. It is a polyglutamine genetic disorder caused by a CAG trinucleotide repeat expansion in the androgen receptor gene on the X-chromosome.7 Degeneration of motor neurons in the spinal cord and brainstem results in a slowly progressive disorder characterised by weakness and atrophy of facial, bulbar and limb muscles without upper motor neuron (UMN) signs (figure 1). Cramps, leg weakness, tremor and orolingual fasciculations (see online supplementary video S1) with bulbar symptoms are the most common presenting symptoms. The syndrome affects only men, although female carriers may experience mild symptoms such as cramps.8 Symptom onset is typically between 30 and 50 years of age, but there is marked variability in age of presentation.9 Weakness is typically noted first in the lower limbs and may be symmetrical or asymmetrical, affecting proximal and/or distal muscles.9 A sensory neuropathy is commonly associated with the syndrome and is usually subclinical. Associated androgen resistance may result in gynaecomastia, testicular atrophy and oligospermia. The diagnosis is confirmed through molecular genetic testing with affected men having >39 CAG repeats.10 Life expectancy may be reduced in selected patients, most commonly due to pneumonia resulting from bulbar dysfunction.9

Spinobulbar muscular atrophy (Kennedy's disease): (A) facial asymmetry due to asymmetrical facial muscle weakness which is accentuated by pursing the lips; (B) tongue wasting resulting in scalloping of the lateral borders and midline furrowing.
supplementary video
Distal hereditary motor neuropathies
The dHMNs share the characteristics of a slowly progressive, length-dependent (ie, distal predominant) pattern of LMN weakness.11 They represent a genetically heterogeneous group with significant variability and overlap in clinical phenotypes for many of the known implicated genes. Most are inherited in an AD pattern, but AR and X-linked inheritance patterns have also been described.12 Onset is often in childhood or teens, but adult onset is not uncommon. Upper limb predominance (dHMN V), vocal cord paralysis (dHMN type VII), respiratory distress (dHMN type VI) and pyramidal signs may be associated features in some patients. Significant sensory involvement is absent, allowing differentiation from axonal forms of Charcot-Marie-Tooth disease, although some mutations may cause both phenotypes.11
Despite significant advances in molecular genetics, a disease-causing mutation is only identified in ∼15% of patients with a typical presentation of dHMN.13 Mutations in the HSPB1, HSPB8 and BSCL2 genes are the most frequent causes of AD dHMN. Mutations in HSPB1 and HSPB8 are associated with a classical length-dependent motor neuropathy beginning in the lower limbs which may present in childhood (dHMN type I) or adulthood (dHMN type II).14 Several phenotypes associated with mutations in BSCL2 have been described and include (1) dHMN type II with a length-dependent motor neuropathy, (2) dHMN V presenting with a predominantly upper limb distal phenotype, (3) dHMN with pyramidal signs and (4) Silver syndrome with atrophy of the intrinsic hand muscles, pyramidal signs and lower limb spasticity.14 The upper limb-onset phenotype (dHMN V) may also result from mutations in GARS with most cases presenting in their second decade with progressive weakness and wasting of the thenar eminence and first dorsal interossei muscles.15 Cramping and pain in the hands on exposure to cold may be an early manifestation.15 The GARS mutation may also present with a classical length-dependent neuropathy beginning in the lower limbs, further highlighting the variability in genotype–phenotype correlations. It remains unclear why mutations in ubiquitously expressed proteins may result in such variable and ‘focal’ phenotypes.11 Bulbar involvement is rare in dHMN, but vocal paralysis secondary to recurrent laryngeal nerve involvement is a feature of dHMN type VII which may result from mutations in dynactin (DCTN1) SLC5A7 or TRPV4.11 AR forms of dHMN are less common but there are increasing numbers of genes being described for this group.11
Although targeted molecular genetic testing was the standard approach to genetic testing in the past, NGS has become a more efficient and cost-effective means of establishing a diagnosis in many settings. This is particularly true with many LMN syndromes as there is a large overlap between genotypes and phenotypes. For example, individual genes can cause phenotypes that have been labelled dHMN, amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegia. Targeted gene panels covering a large number of genes causing these phenotypes are the current preferred NGS diagnostic test. Whole exome and whole genome sequencing which sequence genes encoding proteins, or an individual's entire DNA, respectively, are commonly used in research and increasingly in diagnostic testing and may have particular clinical utility when known suspected genes have been tested by other methods and found to be normal. Such advances in neurogenetics may allow a genetic diagnosis to be established in a greater proportion of patients, but challenges remain including difficulties managing large volumes of data, in the interpretation of sequence variants and determining the pathogenicity of detected mutations. Furthermore, NGS may result in failure to detect certain mutations such as chromosomal deletions or insertions and repeat expansions.
Immune-mediated neuropathies
Several immune-mediated neuropathies may present with a purely motor or motor-predominant neuropathy and can be divided into acute and chronic forms. They are important to differentiate from hereditary and degenerative causes as they may respond to immunotherapy. The acute immune-mediated neuropathies are collectively referred to as Guillain-Barré syndrome (GBS) with a number of variants described including a motor variant known as acute motor axonal neuropathy (AMAN). Chronic forms of the immune-mediated neuropathies include MMN and CIDP.
Guillain-Barré syndrome
GBS may present as a pure motor disorder. Both the classic acute inflammatory demyelinating polyneuropathy (AIDP) form and the recently described AMAN can be pure motor, although the latter is defined by its pure motor phenotype and axonal characteristics. Clinically, AMAN is part of the GBS spectrum but distinguished by normal sensation and nerve conduction studies characterised by low distal motor evoked amplitudes, normal sensory conductions and no features of demyelination. Prognosis is similar to that seen in the AIDP form of GBS unless there is secondary axonal degeneration in which case, like in the AIDP form, recovery is delayed. There is a strong association between AMAN and IgG antibodies against GM1 or GD1a which may be a result of molecular mimicry resulting from antecedent Campylobacter jejuni infection.16
Chronic immune-mediated neuropathies
MMN typically presents with asymmetrical distal weakness and wasting, without sensory impairment which is slowly progressive and has an upper limb predilection17 (figure 2). Weakness may be out of proportion to muscle wasting and involvement of wrist and/or finger extension at onset should prompt consideration of MMN as a potential diagnosis. Positive features such as twitching, cramping and spasm are relatively common in MMN and may be the presenting symptom.18 Bulbar and respiratory involvement are not typical, although respiratory symptoms may occur due to phrenic nerve involvement.

Asymmetric wasting of thenar eminence in a 71-year-old male with an upper limb predominant motor neuropathy associated with anti-GM1 IgM antibody (A). High doses of intravenous immunoglobulin were required to achieve disease stabilisation. The CMAP was unrecordable on the right from APB. The distal APB CMAP on the left was normal, but there was marked dispersion and reduction in CMAP amplitude with stimulation at the elbow (B). A, amplitude; A, area; APB, abductor pollicis brevis; d, duration; CMAP, compound muscle action potential; CV, conduction velocity; NCS, nerve conduction studies.
A definitive diagnosis of MMN requires demonstration of focal motor conduction block on neurophysiological studies with normal sensory nerve conduction across the region of block.19 As conduction block may be difficult to demonstrate and may occur in proximal segments, comprehensive neurophysiology should be performed and proximal stimulation may be required. A normal compound muscle action potential amplitude in a weak muscle with neurogenic recruitment on EMG suggests the presence of conduction block. Anti-GM1 IgM is present in ∼50% of cases with a high titre supporting a diagnosis of MMN.17 MRI may reveal asymmetrical nerve enlargement and increased signal intensity on T2-weighted images of the brachial plexus.20 Ultrasound imaging may show multiple sites of peripheral nerve enlargement in the arms, including segments without conduction abnormalities.21
Intravenous immunoglobulin (IVIg) is the accepted treatment for MMN.17 Dosing must be individualised and no optimal dosing strategy has been established, although high doses of IVIg are often required.22 Furthermore, despite treatment, MMN is often associated with progressive axonal loss and functional decline.17
A purely ‘axonal’ form of MMN has been described which lacks demonstrable partial motor conduction block, demyelinating features and anti-GM1 antibodies, but may respond to IVIg.23 It is important to recognise, however, that at least some of the ‘apparent’ cases of ‘axonal MMN’ may represent MMN with very proximal conduction blocks which are not detectable with standard neurophysiological techniques.24 A trial of IVIg may be warranted in select cases of asymmetrical adult-onset LMN syndromes without demonstrable conduction block, particularly those with distal upper limb-onset weakness.25 A pure motor variant of CIDP with sparing of sensory fibres clinically and neurophysiologically has also been reported. As in MMN, the neuropathy appears to be responsive to IVIg, but not corticosteroids, which may cause deterioration.26
Motor neuron disease
MND is widely recognised as a fatal heterogeneous group of neurodegenerative disorders. The combination of upper and LMN signs is the pathognomonic hallmark with this variant referred to as ALS. Pure UMN and LMN forms have also been described representing opposite clinical ends of the MND spectrum.27
Progressive muscular atrophy
The LMN phenotype of MND (progressive muscular atrophy, PMA) is characterised by progressive LMN signs without clinical evidence of UMN dysfunction, although a significant proportion develop UMN signs during the disease course.28 It is estimated that the syndrome represents ∼5% of MND cases, and may be characterised by slower progression than other forms of MND.28 In the absence of UMN signs, confident differentiation from other LMN syndromes may be difficult, often requiring a period of observation to assess progression. The novel neurophysiological technique of threshold tracking transcranial magnetic stimulation (TMS) has been a major advance allowing for objective assessment of the functional integrity of the UMN system.29 Threshold tracking TMS has demonstrated evidence of cortical hyperexcitability in MND and may play a role in the differentiation from mimic disorders by providing objective evidence of UMN dysfunction when it is not evident clinically.30
Flail arm syndrome
The flail arm syndrome (brachial amyotrophic diplegia or ‘man-in-the-barrel’ syndrome) is a distinct variant of MND characterised by a progressive, predominantly LMN pattern of weakness in the upper limbs, typically beginning in proximal muscle groups with progression to distal involvement. Original descriptions were of a symmetrical pattern of weakness, but there may be some asymmetry, particularly early in the disease course. Mild UMN signs are often present in the lower limbs. There is a striking male predominance with a male-to-female ratio of 4:1,31 and up to 10:1 in some series.32 Prognosis is better than that of classical ALS with a median survival of ∼5 years.31 The flail arm phenotype is associated with cortical hyperexcitability with a similar pattern to that seen in ALS.32 Hence, TMS may be a useful adjunct in differentiating this MND variant from other more benign LMN syndromes with upper limb predominance.
Flail leg syndrome
The flail leg variant of MND (also known as the pseudopolyneuritic variant) is characterised by a progressive, asymmetrical predominantly LMN pattern of weakness with distal-onset weakness and wasting of the lower limbs. UMN signs often emerge over time.31 Progression is slower than classical ALS with a median time of 33 months to involvement of a second region and median survival of almost 6 years. In contrast to the flail arm variant, the flail leg group show an equal male-to-female ratio.31 As with other forms of MND, cortical hyperexcitability is a feature of the flail leg syndrome, but only when UMN signs are present. In contrast, features of cortical hyperexcitability were not demonstrated in patients who lacked UMN signs on clinical examination.33
Familial LMN variants of MND
A variety of genetic mutations may be associated with significant LMN involvement with or without UMN signs. They include mutations in copper–zinc superoxide dismutase type 1 (SOD1), fused in sarcoma (FUS), vesicle-associated membrane protein/synaptobrevin-associated membrane protein B (VAPB) and chromatin-modifying protein 2b (CHMP2B) genes.34 Most of the known forms are inherited in an AD pattern.
SOD1 gene mutations account for 20% of AD familial MND and are the second most common cause of familial MND (following the expanded hexanucleotide repeat in the C9ORF72 gene associated with the ALS-frontotemporal dementia spectrum).27 The A4V missense mutation has been demonstrated to occur in around 40% of patients with SOD1 mutations in North American series and is rare in the European population.35 LMN signs predominate with absent or mild UMN features. Disease progression is particularly rapid with a median survival of 1.2 years from disease onset.35 The A4T mutation is also associated with a similarly rapid disease course and LMN predominant syndrome.36 In contrast, the G93C mutation has been associated with a pure LMN clinical phenotype without bulbar involvement and more favourable prognosis with a median survival of 153 months.37 The D101N mutation in exon 4 of the SOD1 gene has been associated with PMA with limited bulbar involvement and rapid disease course with mean time to death from respiratory failure of 28 months.38
Mutations in the FUS gene account for ∼5% of familial MND and may present with PMA or LMN predominant MND.39 Mutations in the VAPB gene have been associated with a range of phenotypes including PMA and late-onset SMA.40 Mutations in the CHMP2B gene were first linked to frontotemporal dementia, but may be associated with PMA or ALS. In one series, CHMP2B mutations were found in 10% of patients with LMN predominant ALS, although most cases exhibited a sporadic phenotype.41
Monomelic amyotrophy
Monomelic amyotrophy (MMA) is a LMN disorder that presents with insidious onset of focal wasting and weakness, most commonly affecting the upper limb unilaterally, although it can rarely affect a lower extremity. Symptoms typically progress over a period of 1–5 years and then plateau.42 Bulbar, sensory and pyramidal signs are absent. The condition has a striking male predominance with a male-to-female ratio of 10:1.42 It is seen more commonly in Asian countries with a median age of onset in the late teens or early 20s.42
The typical pattern of weakness and wasting in upper extremity MMA (also known as Hirayama disease) is distal predominant affecting the hand and forearm muscles, with C7-T1 innervated muscles classically affected. Preservation of brachioradialis muscle bulk (a C6 innervated muscle) with wasting of C7 innervated forearm muscles may result in the clinical sign described by Hirayama et al43 as ‘oblique amyotrophy’. Symptoms may be aggravated by cold weather and there may be an associated mild tremor on finger extension. Less severe involvement of the contralateral upper limb may occur in a significant proportion of patients.42 MRI findings in Hirayama disease may reveal lower cervical cord atrophy (C5–C7), asymmetric cord flattening and/or intramedullary hyperintensity. Anterior displacement of the dorsal dura on neck flexion may be seen and venous plexus engorgement may give the appearance of an enhancing epidural crescent along the posterior aspect of the cord on neck flexion views.44
Lower extremity MMA presents with weakness and wasting of a unilateral lower limb, although less severe or subclinical involvement of the contralateral limb may occur. It is less common than Hirayama disease but is also characterised by male predominance and benign course. Posterior leg muscles are disproportionately affected with imaging studies demonstrating most severe involvement of gastrocnemius and soleus muscles with marked asymmetry.45 ,46 The degree of wasting may be out of proportion to weakness and disability.45
Segmental lower motor neuron disease
While progression typically arrests within a few years in Hirayama disease, segmental LMN disease is a localised form of sporadic adult-onset LMN disease affecting the upper limbs characterised by progression over a longer period extending up to 20 years.47 The clinical presentation is with asymmetrical LMN signs localised to the upper extremities with unilateral dominance. Both proximal and distal forms are recognised. The clinical course is favourable with progression to generalised MND/ALS rare. MRI may reveal a ‘snake eyes’ appearance with T2-signal hyperintensity in the anterior horns of multiple segments of the cervical cord, although this is a non-specific finding and has been associated with a number of other LMN syndromes including cervical spondylosis and infection48 (figure 3).

This 46-year-old man presented with a 20-year history of progressive distal wasting and weakness of the right hand and forearm muscles. Symptoms developed in the left hand 5 years prior to presentation. Upper limb reflexes were depressed. Needle electromyography revealed chronic neurogenic changes in clinically affected muscles. There were no sensory abnormalities. (A and B) Asymmetrical wasting of the hands and forearm affecting C7-T1 musculature with striking preservation of brachioradialis in the right upper limb; (C) sagittal T2-weighted STIR image demonstrating a linear hyperintensity within the cervical cord at C6 and C7 associated with cord atrophy; (D) axial T2-weighted image with ‘snake eyes’ appearance in the anterior horns. STIR, Short TI Inversion Recovery.
Poliomyelitis and other infections
Poliomyelitis is a rare complication of poliovirus infection characterised by the destruction of the anterior horn cells causing motor weakness. Wild-type polio has largely been eradicated and remains endemic only along the borders of Pakistan and Afghanistan.49 The typical presentation is of acute flaccid paralysis which is asymmetrical and affects the lower limbs more frequently than the upper limbs.50 Weakness is most prominent in proximal muscle groups. Myalgias and muscle spasm are often prominent prior to onset of weakness. Bulbar and respiratory involvement may occur. PCR of the virus from cerebrospinal fluid is the gold standard for confirming the diagnosis. Most affected patients recover strength, although a significant proportion are left with some residual weakness.
Other enteroviruses (including coxsackievirus, echoviruses and enterovirus 71) and the flaviviruses, such as West Nile virus may also cause acute flaccid paralysis due to anterior horn cell disease.50
Postpolio syndrome
Postpolio syndrome develops after a period of stability in a proportion of patients who have recovered from acute poliomyelitis. Symptoms may include the development of new weakness and muscle atrophy, fatigue and/or pain.50 The cause of postpolio syndrome remains unclear and may be due to the degeneration of enlarged reinnervated motor units.
Other LMN syndromes
Although rare, lead and porphyric neuropathies are briefly discussed here, as they are treatable causes of motor neuropathies. Lead toxicity can lead to a subacute motor neuropathy which classically affects the wrist and finger extensors before spreading to other muscles and hence may be confused with MMN.51 Porphyria, an inherited metabolic disorder of heme biosynthesis, may present with an acute or subacute predominantly motor neuropathy also with focal weakness at onset, such as wristdrop or footdrop. The acute onset may lead to confusion with AMAN. Both lead and porphyric neuropathies are typically associated with involvement of other organ systems and additional features may include gastrointestinal symptoms, cognitive disturbance and haematological changes.51 ,52 A family history of symptoms of porphyria or history of occupational exposure to lead may provide clues to the diagnosis. Lead toxicity is treatable with chelation and porphyria with haematin.
Key differentiating features
The LMN syndromes represent a group of conditions with a variety of presentations and varied underlying disease mechanisms. While a thorough clinical assessment and neurophysiology can usually delineate or assist in classification of the different syndromes, peripheral nerve and muscle imaging and the rapidly advancing field of NGS are additional diagnostic tools available to clinicians. Neurophysiological findings in the majority of LMN syndromes consist of axonal loss with a reduction in compound muscle action potential amplitudes with normal or slightly reduced motor conduction velocities. In contrast, findings suggestive of an immune-mediated motor neuropathy include features such as significantly reduced conduction velocities, focal partial motor conduction block and prolongation of F-wave latencies. While treatments are currently limited for most syndromes, immune-mediated LMN disorders may respond to immunotherapy and hence are important to distinguish from hereditary and degenerative causes.
Figure 4 outlines a diagnostic approach for the patient presenting with a LMN syndrome. Table 1 outlines the typical clinical features of the different LMN syndromes. An acute presentation of a LMN syndrome with onset over days to weeks should prompt consideration of an immune, toxic, metabolic or infective aetiology. The major differentials for a chronic monomelic or asymmetrical presentation include MND, MMN and MMA. Early involvement of finger and wrist extensors, focal motor conduction block on neurophysiology and/or the presence of anti-GM1 IgM antibody should prompt consideration of MMN as the diagnosis and warrants a treatment trial of IVIg. Positive symptoms, such as cramping and spasm are common in MMN. In contrast, positive symptoms are rarely a prominent presenting symptom in MND with negative features of LMN dysfunction dominating.53
Clinical features of LMN syndromes

Diagnostic algorithm for a patient presenting with a LMN syndrome. ALS, amyotrophic lateral sclerosis; CB, conduction block; dHMN, distal hereditary motor neuropathy; FHx, family history; GBS, Guillain-Barré syndrome; LL, lower limb; LMN, lower motor neuron; MMA, monomelic amyotrophy; MMN, multifocal motor neuropathy; MND, motor neuron disease; NCS, nerve conduction studies; NGS, next-generation sequencing; PMA, progressive muscular atrophy; SBMA, spinobulbar muscular atrophy; SMA, spinal muscular atrophy; UL, upper limb.
Monomelic weakness, particularly in a young man, with progression over several years followed by stabilisation may be indicative of MMA, particularly if the sign of oblique amyotrophy or typical MRI changes are present. The clinical distinction between MMA and MND may be difficult early in the disease course, and tends to be determined by an extended period of clinical observation. It has been suggested that progression to MND should only be excluded if there has been no progression beyond the upper limb within 3 years.54 A similar timeframe could also be applied to lower extremity MMA. The presence of cortical hyperexcitability on threshold tracking TMS may be a helpful adjunct in differentiating LMN-predominant MND from other LMN disorders although the results must be interpreted in the context of other clinical and neurophysiological findings. Furthermore, EMG may reveal features of active denervation and/or reinnervation changes in clinically normal limbs in MND.
Although not always present, enquiry regarding recent or past pain should be made as it may suggest compressive or inflammatory causes, including spondylosis, brachial and lumbosacral plexitis and neuritis. As cervical and lumbar spondylosis are common conditions, imaging of the spine and brachial/lumbosacral plexus should always be considered, particularly in patients with asymmetrical involvement to exclude radiculopathy and to assess for nerve root/plexus pathology which may suggest an immune aetiology. Cervical spine MRI with neck flexion views should be considered when Hirayama's disease is a differential diagnosis.
A hereditary aetiology may be suspected by a history of slowly progressive weakness with onset in childhood or early adulthood and/or the presence of a positive family history. An insidious onset of slowly progressive relatively symmetrical weakness over many years is suggestive of dHMN when the pattern of weakness is distal or SMA if weakness is predominantly proximal. Upper limb predominance may indicate dHMN with a mutation in GARS or BSCL2. Symmetrical weakness confined to the upper limbs may indicate the flail arm variant of MND, but here the weakness typically begins in proximal muscle groups and extends distally with a relatively progressive course. Slowly progressive asymmetrical weakness over many years in a segmental pattern should prompt consideration of segmental LMN disease. Bulbar involvement may occur in MND and SMA but does not occur in MMA and is not typical in MMN or dHMN.
Pathophysiology
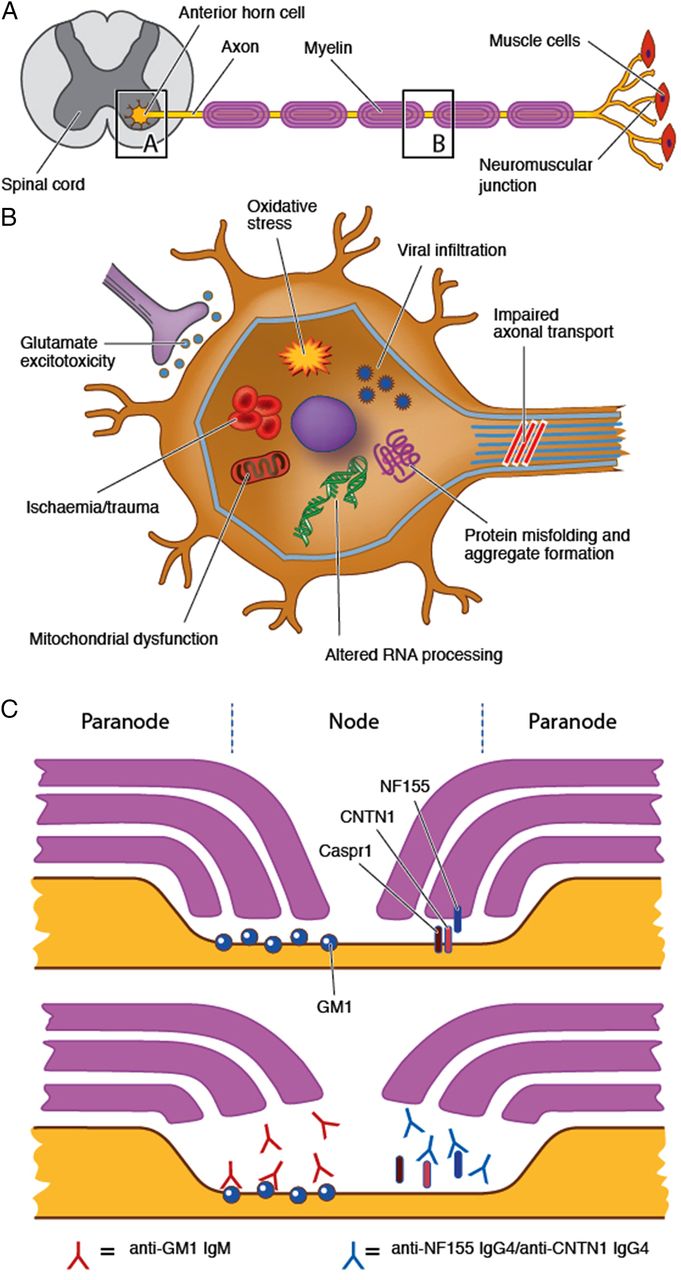
Pathogenic mechanisms of axonal degeneration and cell death in the genetic LMN syndromes are a complex interaction of multiple factors (figure 5). Implicated mechanisms in dHMN include protein misfolding and aggregate formation as well as impaired axonal transport and RNA processing.55 Mutations in the SMN1 gene causing SMA result in reduction in intracellular survival motor neuron (SMN) protein levels. This is believed to cause death of motor neurons in the spinal cord and lower brainstem by interfering with RNA processing and spliceosome assembly.56

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed pathogenic mechanisms for LMN syndromes. LMN syndromes may arise from disease processes affecting the anterior horn cell or the motor axon and/or its surrounding myelin. (A) A variety of mechanisms have been implicated in the degenerative and hereditary syndromes including mitochondrial dysfunction, altered RNA processing and impaired axonal transport (see text for further details). (B) Anti-GM1 antibodies may bind to GM1 in the paranodal region leading to disruption of ion channel clusters and paranodal anatomy. Although not a purely LMN syndrome, IgG4 antibodies against NF155 and CNTN1 have recently been described and may similarly disrupt paranodal anatomy resulting in a sensorimotor neuropathy. CNTN1, contactin-1; LMN, lower motor neuron; NF155, neurofascin-155.
Neurodegeneration in MND is postulated to occur through multifactorial mechanisms including glutamate excitotoxicity, axonal transport dysfunction, RNA processing defects, mitochondrial dysfunction and oxidative stress.27 ,57 ,58 SOD1 mutations lead to abnormalities in protein degradation, resulting in aggregate formation and triggering damage to axonal transport, mitochondrial function and a variety of other cellular functions.57 However, the precise mechanisms underlying LMN-dominant presentations of MND remain unknown.
Similarly, the aetiology of LMN disorders with restricted regions of involvement remains unclear. Pathological studies have demonstrated focal degeneration restricted to anterior horn cells in MMA.43 It has been postulated that upper extremity MMA may be a myelopathy related to flexion movements of the neck with resultant ischaemic damage to the anterior horn cells of the cervical cord.44 Alternatively MMA may represent a primary localised neurodegenerative disorder of the anterior horn cell.
Pathophysiological mechanisms in immune-mediated neuropathies reflect a different aetiology, involving aberrant cellular and humoral immune responses. The site of pathology in MMN is likely to lie in the region of the node of Ranvier and paranode. It has been postulated that anti-GM1 antibodies may bind to the ganglioside GM1 which is enriched in the paranodal region, activating complement and leading to the disruption of ion channel clusters and paranodal anatomy.17 ,59 It has been suggested that GM1 is more abundant within motor than in sensory nerves, resulting in the preferential motor selectivity seen in MMN, although late sensory involvement has been described.17 In contrast to MMN, antigenic targets in CIDP remain largely elusive, although IgG4 antibodies against the paranodal proteins neurofascin-155 and contactin-1 have recently been described and are associated with a severe sensory and motor CIDP phenotype with poor response to IVIg, but favourable response to rituximab.60 ,61 Further delineation of pathophysiological mechanisms underlying different LMN syndromes will contribute to improved disease classification and the development of targeted treatments.
Conclusion
LMN syndromes represent a spectrum of disorders with a variety of underlying aetiologies and presentations. Clinical assessment combined with neurophysiology are often adequate to establish a diagnosis, but advances in genetic and imaging techniques are further diagnostic tools becoming increasingly accessible to clinicians. Although establishing an accurate diagnosis in LMN presentations can be clinically challenging at times, it is important due to prognostic and treatment implications.
References
Footnotes
Contributors MCK, SBP and NG conceived the idea for the article. NG drafted the manuscript. All authors revised the manuscript critically for important intellectual content, and gave final approval of the version to be published.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed.