Article Text

Abstract
Objective We characterised the clinical course, treatment and outcomes in 59 patients with relapsing myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination.
Methods We evaluated clinical phenotypes, annualised relapse rates (ARR) prior and on immunotherapy and Expanded Disability Status Scale (EDSS), in 218 demyelinating episodes from 33 paediatric and 26 adult patients.
Results The most common initial presentation in the cohort was optic neuritis (ON) in 54% (bilateral (BON) 32%, unilateral (UON) 22%), followed by acute disseminated encephalomyelitis (ADEM) (20%), which occurred exclusively in children. ON was the dominant phenotype (UON 35%, BON 19%) of all clinical episodes. 109/226 (48%) MRIs had no brain lesions. Patients were steroid responsive, but 70% of episodes treated with oral prednisone relapsed, particularly at doses <10 mg daily or within 2 months of cessation. Immunotherapy, including maintenance prednisone (P=0.0004), intravenous immunoglobulin, rituximab and mycophenolate, all reduced median ARRs on-treatment. Treatment failure rates were lower in patients on maintenance steroids (5%) compared with non-steroidal maintenance immunotherapy (38%) (P=0.016). 58% of patients experienced residual disability (average follow-up 61 months, visual loss in 24%). Patients with ON were less likely to have sustained disability defined by a final EDSS of ≥2 (OR 0.15, P=0.032), while those who had any myelitis were more likely to have sustained residual deficits (OR 3.56, P=0.077).
Conclusion Relapsing MOG antibody-associated demyelination is strongly associated with ON across all age groups and ADEM in children. Patients are highly responsive to steroids, but vulnerable to relapse on steroid reduction and cessation.
- myelin oligodendrocyte glycoprotein antibodies
- optic neuritis
- acute disseminated encephalomyelitis
- therapy
- outcomes
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- myelin oligodendrocyte glycoprotein antibodies
- optic neuritis
- acute disseminated encephalomyelitis
- therapy
- outcomes
Introduction
Myelin oligodendrocyte glycoprotein (MOG) is a component of myelin expressed exclusively in the central nervous system (CNS).1 2 Using cell-based assays that preserve the conformational structure of full-length human MOG, antibodies targeting MOG have been recently identified in both children and adults with demyelination including acute disseminated encephalomyelitis (ADEM), optic neuritis (ON) and transverse myelitis (TM), with some patients fulfilling revised diagnostic criteria for aquaporin-4 (AQP4) antibody-negative neuromyelitis optica spectrum disorder (NMOSD).3–8 There is increasing evidence that some patients have a relapsing course.6 9–11
Accurate diagnosis of the demyelinating aetiology is essential as treatment options vary between multiple sclerosis (MS) and NMOSD. While MS is frequently treated with disease-modifying immunomodulatory therapy, some agents used in MS such as interferon, natalizumab and fingolimod have been shown to not benefit or have a detrimental impact in AQP4 antibody-positive NMOSD.12–14 Immunosuppressants such as azathioprine, mycophenolate and rituximab all reduce relapse rates in AQP4 antibody-positive NMOSD.15–17 Data regarding the efficacy of immunotherapy in patients with MOG antibodies is limited. Herein, we detail the response to specific immunotherapies in a large multicentre cohort of paediatric and adult patients with relapsing MOG antibody-associated demyelination.
Methods
Study design, patient recruitment and antibody testing
Patients were classified as paediatric (≤16) or adult (>16) based on age at disease onset. All serum samples were tested for MOG antibodies in a single centre using a flow cytometry live cell-based assay as previously described.6 10 We identified 61 patients with relapsing demyelination (≥2 episodes) for whom detailed clinical information was available, from neurology, neuroimmunology and neuro-ophthalmology clinics in 25 centres in Australasia (Australia n=22, New Zealand n=2, Singapore n=1). An acute episode was defined as a new neurological deficit lasting at least 24 hours.17 A relapse was any new CNS symptom/sign lasting >24 hours in the absence of other causes, and supported clinically or radiologically.15 This included neurological symptoms/signs that occurred during the course of treatment weaning, but were distinct from the initial presentation and separated by a period of recovery/improvement. Two paediatric patients were excluded due to being lost to follow-up. In total, 59 patients (paediatric n=33, adult n=26) were included. Data was retrospectively collected by SR (adult) and RCD (paediatric) through telephone interviews with each patient’s neurologist, using a structured questionnaire created for this study (see online supplementary appendix 1). All patients had a minimum of 12 months of follow-up.
Supplementary file 1
Therapeutic efficacy
We calculated annualised relapse rates (ARR) for each patient as the number of relapses/year pretreatment and on-treatment.18 19 Pretreatment ARRs were calculated after excluding the index episode, to avoid an inaccurately high value.18 19 All ARRs were calculated only if the period of time being assessed was ≥6 months. We calculated the cumulative cohort ARR for each treatment (total number of relapses in cohort/total patient-years on each treatment). Patients were included in calculations of ARR if they fulfilled the following criteria.
Corticosteroids
Pulsed intravenous methylprednisolone (dose 20–30 mg/kg/day for children, 500 mg–1 g/day for adults, over 3–5 days) and/or a taper of oral prednisone (starting dose 1–2 mg/kg/day in children and 1 mg/kg in adults) with variable tapering durations, based on individual clinician preference. Patients were classified as being on ‘maintenance corticosteroids’ if they were given oral corticosteroids after second or further demyelinating episodes at daily doses >10 mg for patients >40 kg in weight, and >5 mg for patients ≤40 kg in weight.
Intravenous immunoglobulin
An induction course dose of 2 g/kg, and subsequent monthly doses of 1 g/kg/infusion.
Steroid-sparing immunosuppression
Treatment with mycophenolate, azathioprine or methotrexate at adequate doses for weight.15
Rituximab
ARR was calculated for 6 months after rituximab infusion. Rituximab failure was defined as a relapse occurring between 2 weeks and 6 months after treatment. A ‘repopulation relapse’ was a relapse associated with B-cell repopulation (CD19 count ≥10×106 cells/L).19
Outcomes
Patient outcomes were characterised using the Expanded Disability Status Scale (EDSS).20 Recovery from each demyelinating episode was additionally classified as complete, partial or no recovery. Complete recovery included a normal neurological examination. Residual deficits were classified including visual impairment (with reduced visual acuity), sensory, motor, cognitive and/or sphincter dysfunction and epilepsy.
Standard protocol approval and patient consent
Ethics and research protocol approval (NEAF 12/SCHN/395) was granted by the individual ethics committees for all participating hospitals. Informed consent was obtained from all patients.
Statistical analyses
Patient and clinical characteristics were described by frequencies and percentages for categorical variables; and mean, median and range for continuous variables. Predictors of categorical outcomes were examined using Χ2 tests, as well as univariable and multivariable logistic regression models and described by OR and 95% CIs. Comparisons between groups of patients were performed using Χ2 tests and Wilcoxon rank-sum or Kruskal-Wallis tests. ARRs were tested using the Wilcoxon signed rank test to examine whether ARRs decreased during treatment. Treatment failure rates were described with 95% CIs using the Wilson method. Comparison in treatment failure rates between maintenance steroids and non-steroidal immunosuppression was performed using Fisher’s exact test. Figures were generated using Prism Software V.6.0 (GraphPad Software, La Jolla, California, USA) and Adobe Illustrator CS6 (Adobe Systems, San Jose, California, USA).
Results
Clinical characterisation
We identified 59 patients (40 female, 19 male) with relapsing MOG antibody-associated demyelination, with a total of 218 episodes. Patients were followed up for an average of 61 months (median 45, range 12–288) (table 1, online supplementary tables 1 and 2).
Cohort summary of relapsing paediatric and adult MOG antibody-positive patients: clinical and laboratory characteristics
Supplementary file 5
The median age of onset for the total cohort was 12 years (mean 21 range 1–74), in children aged 6 years (mean 7, range 1–16), and in adults aged 37 years (mean 40, range 18–74). Forty-three out of fifty-nine (73%) patients were Caucasian. Forty-seven per cent had an infectious prodrome (including myalgia, fevers or preceding symptoms consistent with respiratory infection) in the month prior to their first episode. Two patients had a vaccination within 3 weeks prior to an episode. Three patients had their first presentation (n=2) or relapse (n=1) ≤4 months post partum. Four patients had coexistent autoimmunity (type 1 diabetes mellitus and Hashimoto’s thyroiditis, elevated thyroid antibodies, Henoch-Schönlein purpura and prior LGI1 antibody-positive encephalitis). Twenty out of fifty-nine patients had two demyelinating episodes, and 39/59 had ≥3 (see online supplementary figure 1A and appendix 2).
There was a difference in clinical phenotypes on first presentation between children and adults (P=0.006). The most common presenting phenotype in the total cohort was ON (54%) (bilateral ON (BON) 32%, unilateral ON (UON) 22%) (figure 1A). ADEM (2013 criteria)21 was the most common presentation in children (36%), followed by BON (24%) and UON (15%). Adults presented primarily with ON 73% (BON 42%, UON 31%). TM was less common. Isolated seizures with mild encephalopathy and subtle inflammatory changes in CSF or radiology, but in the absence of clinical and radiological features of ADEM,21 were present in two children. Mixed presentations were common (see online supplementary table 3). Seven out of fifty-nine patients (five adults and two children) had simultaneous ON+longitudinally extensive transverse myelitis episodes consistent with the classical description of Devic’s disease.

The clinical phenotype and steroid response in relapsing MOG antibody-associated demyelination. (A) Illustration of the clinical phenotype at first presentation of all patients in the cohort, with a breakdown reflecting the distribution of each clinical phenotype in the paediatric age group (0–16 years at disease onset), and adult group (>16 years at disease onset). (B) Illustration of the clinical phenotype at all presentations (n=218, initial presentation plus relapses) in the cohort, with a breakdown reflecting the distribution of each clinical phenotype in the paediatric and adult group. (C) An illustration of the number of relapses that occurred during tapering oral prednisone (and the dose of prednisone at which these relapses occurred), or following the cessation of an oral prednisone taper (and the time frame at which these relapses occurred). ADEM, acute disseminated encephalomyelitis; BON, bilateral optic neuritis; LETM, longitudinally extensive transverse myelitis; TM, transverse myelitis; UON, unilateral optic neuritis.
When evaluating the clinical phenotype of all 218 episodes in the cohort, ON remained dominant (figure 1B). UON (35%) was most common, followed by BON (19%). ADEM was common in children (15%), but rare in adults (2%). TM was less common in the whole cohort (11%), and more prevalent in adults.
Relapsing ON was the main relapsing clinical syndrome in the total cohort (29%), followed by NMOSD (2015 criteria)8 (25%) (table 1). ADEM was present at some point in the disease course in 31% of the whole cohort; including 52% of children and 4% of adults, which was statistically different (p<0.0001). Fifty-one out of fifty-nine patients had one or more episodes involving ON. Funduscopy results were available in 30/51 patients and 27/30 (90%) had visible optic nerve head swelling with the ON (9/11 (82%) children and 18/19 (95%) adults).
This study identified some uncommon clinical phenotypes associated with MOG antibodies. Some patients presented with sensory symptoms (predominantly lower limb paraesthesia) that were steroid responsive, in the absence of radiological myelitis. One adult patient with relapsing BON presented with recurrent subacute cognitive decline that was reversible with steroids and immunotherapy. It is conceivable that she had concurrent autoimmune encephalitis; however, testing for neuronal cell surface antibodies was negative, and she did not have seizures.
Cerebrospinal fluid analysis
CSF at initial presentation was available in 49/59 patients; 28/48 (58%) had a mononuclear lymphocytic pleocytosis (≥5 mononuclear cellsx106/L) but only 18/49 (37%) had an elevated protein level (>0.40 g/L for children, >0.45 g/L for adults). Forty-five patients were tested for intrathecal oligoclonal bands and only 5/45 (11%) were positive (table 1); 14/49 patients had normal routine CSF analysis.
MRI findings
MRI brain scans were reviewed to confirm whether the patients fulfilled 2010 revised McDonald criteria radiologically for dissemination in space and time at the time of each scan.22 One hundred thirty-one MRIs were performed within 1 month of a demyelinating episode, and 58/131 (44%) of these demonstrated no brain lesions; 54/58 of normal brain scans were performed during isolated ON and/or TM. Only 11/131 acute brain MRIs fulfilled radiological McDonald criteria in seven patients. Ninety-six brain MRIs were performed during an asymptomatic recovery period (≥3 months following presentation); 52/96 (54%) of asymptomatic MRIs had no brain lesions; 37/96 (39%) had residual changes and only 7/96 (7%) had new asymptomatic lesions. These seven MRIs belonged to six patients, and only one out of six patients fulfilled revised McDonald criteria for MS. Two of these six patients had follow-up scans, which showed no new lesions and partial (n=1) or complete (n=1) resolution of previous lesions. Four out of six patients are yet to have further follow-up radiology and have been clinically asymptomatic.
Acute corticosteroid usage and risk of relapse
Treatment data were available for 191/218 episodes; 181/191 episodes were treated with corticosteroids; 35/181 episodes were treated with intravenous methylprednisolone alone and 146/181 episodes were treated with oral steroids (following intravenous methylprednisolone, n=116; or solely as an oral taper, n=30). The median duration of the oral taper was 1.62 months. Following the 146 episodes treated with oral prednisolone taper, there were 103 subsequent episodes, most of which occurred towards the end of the taper (n=41 episodes; mean dose of oral prednisone at relapse 11.5 mg, median 10 mg, range 1.25–60 mg), or shortly after prednisone cessation (n=62 episodes; median duration after prednisone cessation 2 months, range 0–62) (figure 1C). The median length of a planned steroid taper was 1.5 months (mean 3, range 0.25–24) for all episodes followed by a relapse, compared with 5 months (mean 6.9, range 0.75–26) for all episodes not followed by a relapse.
Maintenance treatment efficacy
The ARR pretreatment, on-treatment and post-treatment; cumulative cohort ARR and failure rates for each therapeutic agent are summarised in table 2 (all therapy trialled in each patient is detailed in online supplementary table 4). Only treatments with ≥5 patients who fulfilled inclusion criteria are included. All treatments were associated with a reduction in ARR on-treatment, with maintenance prednisone having the lowest treatment failure rate.
Supplementary file 6
Evaluation of pretreatment and on-treatment ARRs for the main treatment modalities
Twenty patients received maintenance oral prednisone for ≥6 months and were included in ARR calculations (figure 2A, table 2); 1/20 (5%) experienced treatment failure.

Disease activity of myelin oligodendrocyte glycoprotein antibody-positive patients on immunosuppressive therapy. (A) Depictions of the clinical course and disease activity in patients who underwent therapy with maintenance prednisone. In patients who had more than two courses of maintenance prednisone lasting ≥6 months each, the green lines depict the duration of each course of maintenance corticosteroid therapy. Depictions of the clinical course and disease activity in patients who underwent therapy with monthly intravenous immunoglobulin (IVIg) (B), mycophenolate (C) and rituximab (D). (E) Illustration of response to switched therapy in the event of side effects (patient 46 azathioprine to mycophenolate), treatment failure of the initial agent (patient 38 mycophenolate to rituximab, patient 58 methotrexate to rituximab, patient 39 methotrexate to mycophenolate, patient 8 mycophenolate to monthly IVIg and patient 57 azathioprine to mycophenolate), or after exceeding the maximum recommended cumulative dose of the first agent (patient 33 cyclophosphamide to azathioprine). All therapy (or switched therapy) commenced at time point 0. The magenta lines represent a demyelinating episode. The purple squares represent time of switched treatment cessation. The orange circles represent time of latest clinical follow-up. The blue arrows represent timing of rituximab dosing.
Seven out of fourteen patients given IVIg fulfilled inclusion criteria for maintenance therapy (figure 2B). Three patients had six relapses while on adequate treatment with IVIg. Three out of seven (43%) experienced treatment failure. Three relapses in two patients occurred while weaning IVIg doses (n=2) or increasing dosing intervals (n=1).
Sixteen out of seventeen patients given mycophenolate fulfilled inclusion criteria for maintenance therapy (figure 2C), with a median duration of treatment of 15 months (range 6–37 months). Eight patients experienced 10 relapses. Seven out of sixteen (44%) experienced treatment failure. Four out of ten relapses occurred during concomitant prednisone taper. Seven out of ten relapses occurred within the first year of mycophenolate treatment. One patient relapsed 1 month following mycophenolate cessation.
Six out of eight patients given rituximab fulfilled inclusion criteria (figure 2D). Three out of six patients were redosed (four courses in one patient, and three courses each for two patients). Treatment failure occurred in one out of six (17%) patients with two relapses (1 and 3 months after dosing) despite B-cell depletion. Three patients had B-cell repopulation relapses >6 months after treatment and were redosed with rituximab. Two out of six patients remained relapse free following the first infusion of rituximab and have not been retreated.
Treatment failure rates were lower in patients on maintenance steroids (1/20, 5%) compared with patients on trials of non-steroidal maintenance immunosuppression (11/29, 38%) (including mycophenolate (7/16), IVIg (3/7) and rituximab (1/6) (P=0.016)).
Seven out of fifty-nine patients switched maintenance immunosuppression treatment (figure 2E) due to concerns of cumulative dose toxicity in one (cyclophosphamide), intolerable side effects of nausea and vomiting in one (azathioprine) and treatment failure of the initial agent in five (mycophenolate n=2, methotrexate n=2, azathioprine n=1) patients. Two patients experienced a relapse following a switch in treatment, with one out of seven (14%) experiencing treatment failure.
Seven patients were treated with azathioprine. Three patients experienced intolerance with prominent nausea, prompting cessation. Four patients received azathioprine for ≥6 months and two out of four were treatment failures (see online supplementary figure 1B and appendix 2). Two patients were treated with methotrexate. One out of two patients reported nausea and fatigue but continued treatment. Both patients experienced breakthrough relapses despite adequate dose and duration of treatment. Another two patients were treated with cyclophosphamide, with one experiencing treatment failure (see online supplementary table 4). Six out of fifty-nine patients received plasma exchange during treatment, with five patients receiving five exchanges following a single demyelinating episode. Only one patient (patient 34) received monthly plasma exchange over a 24-month period, but has required concomitant steroids, mycophenolate and rituximab to control disease activity. Seven out of fifty-nine patients received treatment with one or more immunomodulatory agents for a presumed diagnosis of MS at some point in their disease course (interferons n=6, glatiramer acetate n=3) (see online supplementary table 4).
Risk factors for poor outcomes and residual disability
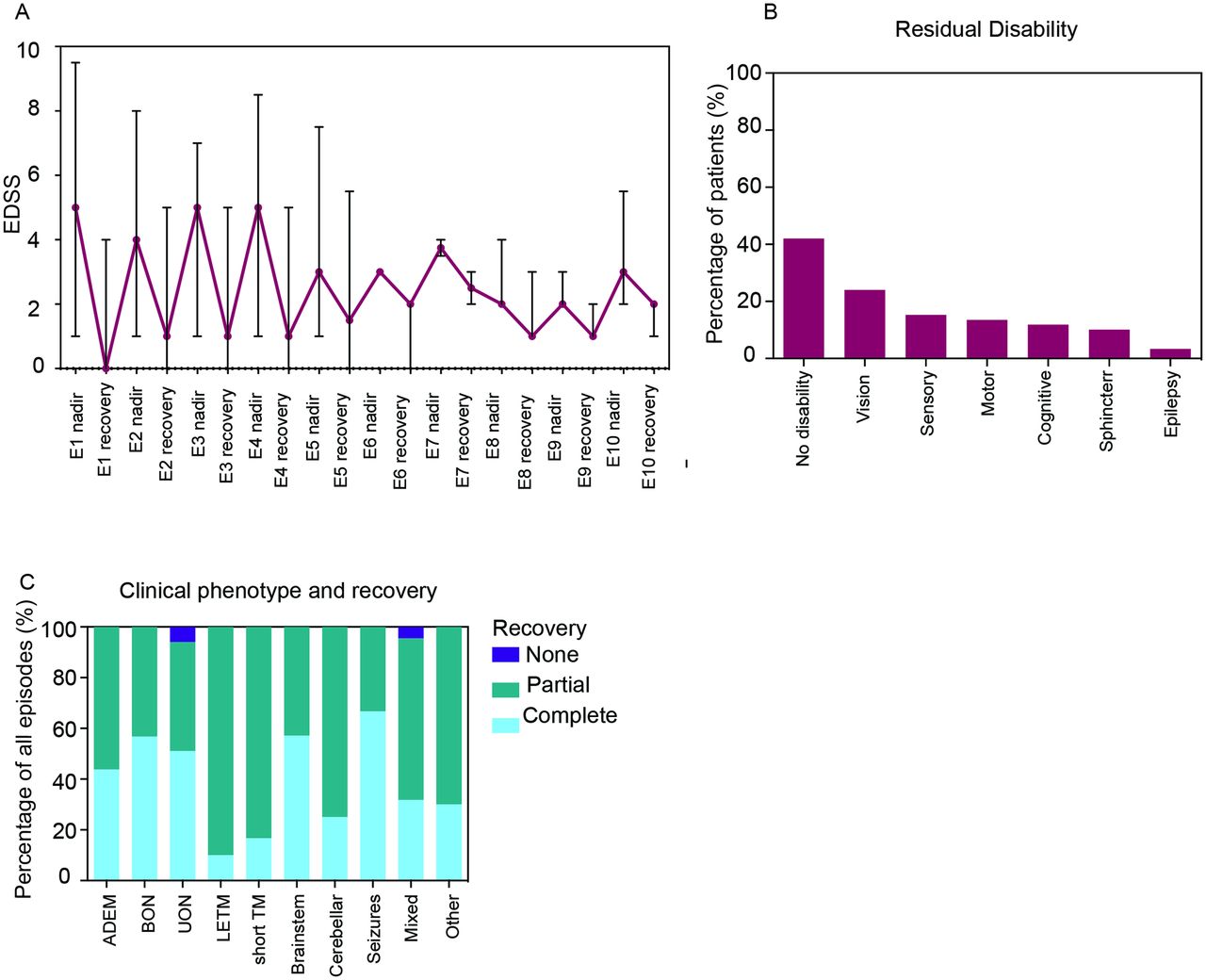
The average EDSS at initial presentation was higher in children (5.6 (median 5, range 2–9.5)) than in adults (4.4 (median 4, range 1–8.5)) (P=0.025), but was similar between the two age groups at latest follow-up (total cohort: 1.3 (median 1, range 0–5.5)) (table 1). The median EDSS at recovery for later episodes was higher than the median EDSS at recovery for earlier episodes (figure 3A, online supplementary table 5). None of the patients who experienced ≥7 episodes returned to an EDSS of 0, suggesting cumulative disability. Logistic regression demonstrated that patients with any ON were less likely to have a final EDSS of ≥2 (OR 0.15, 95% CI 0.03 to 0.85, P=0.032), while those who had any TM were more likely to have sustained residual deficits (OR 3.56, 95% CI 0.87 to 14.46, P=0.077) (table 3).
Supplementary file 7
Risk factors associated with EDSS ≥2 at final follow-up
{kind=link}
{kind=link}
{kind=link}
Recovery and outcomes in relapsing myelin oligodendrocyte glycoprotein antibody-associated demyelination. (A) Median EDSS and range of the cohort at the nadir and at recovery after each demyelinating episode. (B) Percentage of patients with residual disability in a particular domain. (C) Recovery status of all clinical episodes based on clinical phenotype. ADEM, acute disseminated encephalomyelitis; BON, bilateral optic neuritis; E, episode; EDSS, Expanded Disability Status Scale; cyclo, cyclophosphamide; IVIg, intravenous immunoglobulin; LETM, longitudinally extensive transverse myelitis; PLEX, plasma exchange; RTX, rituximab; TM, transverse myelitis; UON, unilateral optic neuritis.
Forty-two per cent (25/59) of patients had no residual disability (figure 3B). The most common deficit was visual acuity loss (14/59, 24%). A further 9/59 had optic disc pallor on funduscopy, without visual acuity reduction. Other disability including sensory (9/59, 15%), motor (8/59, 14%), cognitive (7/59, 12%) and sphincter (6/59, 10%) dysfunction was also present. Epilepsy was rare (2/59, 3%) and limited to two children with seizures as a prominent part of their early presentation who went on to develop refractory seizures, which were difficult to control even with multiple antiepileptic agents. Recovery status was available in 190/218 episodes (figure 3C). The proportion of episodes with complete recovery was higher when the presenting phenotype was ADEM, BON and UON, and lower with TM (see online supplementary table 6).
Supplementary file 8
Discussion
This study identified a female predominance, and over 70% of patients in this Australasian cohort were Caucasian. Coexistent autoimmunity in this cohort was lower than reported for AQP4 antibody-positive patients. Demyelinating episodes in almost half the patients were preceded by an infectious prodrome. BON was the most common initial presentation and UON was the most frequent phenotype throughout the disease course. Optic disc swelling was common as we have previously reported,6 23 and suggestive of MOG antibodies. ADEM was prominent in children,24 while TM was more common in adults. It is additionally likely that a proportion of children previously classified as multiphasic DEM (MDEM) have MOG antibodies.25 26 Brainstem presentations classically recognised in AQP4 antibody-positive disease,27 were present in some patients and have also been recently reported.28 MOG antibody-associated seizures have been described in a few adults,29 and we identified two such children. Only a minority of patients in our cohort fulfilled revised McDonald criteria for MS. Other paediatric cohorts have similarly identified subgroups of MOG antibody-positive children with relapsing demyelination including AQP4 antibody-negative NMOSD, MDEM and recurrent ON.30 Sepulveda et al demonstrated that only a quarter of adult patients they studied were diagnosed as NMOSD, with the majority of their MOG antibody-positive patients having isolated optic neuritis, and a much lower frequency having TM, which were similar to the findings in our cohort.31 Interestingly, a recent study32 identified a few patients with MOG antibodies who had clinically definite MS and fulfilled revised McDonald criteria. These patients had brainstem and spinal cord involvement with a severe disease course, numerous relapses, failure of immunomodulatory therapy and a favourable response to plasma exchange in some. This finding emphasises that there remains diversity in phenotypes associated with MOG antibody-associated demyelination, and that some overlap may be present between patients with clinically definite MS and MOG antibodies.
A large proportion of patient MRIs in this study showed no brain lesions, and only a small percentage fulfilled revised McDonald criteria. This contrasts with MS radiology, and may be helpful diagnostically. Optic nerve and brain radiology may discriminate between MOG antibody-associated demyelination, AQP4 antibody-positive NMOSD and MS.23 33 Indeed, a recent study has identified that the presence of large ‘fluffy’ lesions or three or fewer brain lesions is suggestive of MOG antibody-associated demyelination.34
This study identified that patients with relapsing MOG antibody-associated demyelination are steroid responsive, but frequently relapse at daily prednisone doses <10 mg, within the first 2 months of steroid cessation, and with a rapid taper. This data provides further detail to corroborate previous reports on poststeroid relapses by our group6 as well as a European cohort.11 A number of patients had an extremely rapid return to baseline within 48 hours following steroid initiation (personal observation). Some patients on maintenance low-dose prednisone alone had a relapse-free course, suggesting this is effective in sustaining remission, but the significant long-term metabolic and bone health-related adverse effects warrant caution. A subgroup of MOG antibody-positive patients remained relapse free on no immunotherapy for a long time after initial treatment with steroids, and had a relapse many years later.
This study identified improvement in ARRs with most immunotherapies evaluated. Treatment failure rates were lowest with maintenance oral prednisone and rituximab. Weaning doses of IVIg and prednisone as well as B-cell repopulation after rituximab frequently resulted in relapses, further emphasising the effect of immune therapy in relapse prevention in this condition. In relapsing MOG antibody-positive patients needing rituximab, regular CD19 monitoring and proactively redosing a brittle patient in the event of B-cell repopulation might reduce the incidence of repopulation relapses, as demonstrated in NMOSD.35 36 A recent study evaluating 67 patients with NMOSD suggested mycophenolate showed efficacy and was well tolerated in patients with NMOSD regardless of AQP4 antibody status; however, this cohort only included five MOG antibody-positive patients.37 In our cohort, mycophenolate also appeared to have effect, but treatment failure rates were higher and relapses were often associated with steroid taper suggesting the steroid was producing the benefit in these patients. Switching maintenance immunotherapy following treatment failure on the initial agent significantly reduced relapse rates, supporting the need for flexibility in management.
MOG antibody-associated demyelination has been suggested to have a more favourable prognosis compared with AQP4 antibody-associated disease.5 7 38 39 The median EDSS at acute presentation in this relapsing cohort was 5 and at latest clinical follow-up was 1, suggesting that the majority of patients did have a good outcome. We showed that the severity of the first episode appeared to be worse than relapses—this could be due to either patients presenting earlier or being on immunotherapy at the time of relapse, or potentially due to the nature of the disease process. Interestingly, having ON at some point in the disease course appeared to be protective with regard to sustained disability. In contrast, there was a trend in patients with TM towards having more severe residual deficits. We showed only 42% of patients in the total cohort had no residual disability, suggesting that EDSS measures may underestimate non-motor deficits, and that residual disability in this condition is common. A recent study, mainly in adults, by Jarius et al 11 similarly highlights the potential for serious disability in these patients. While some studies show early evidence that persistent MOG antibody seropositivity may be associated with relapsing disease,40 larger numbers of patients followed up for longer periods of time are required to clarify the therapeutic and prognostic implications of MOG antibody seropositivity in a clinical setting.
Our data suggests that in relapsing MOG antibody-positive patients, a prolonged steroid taper may reduce the chance of early relapses and provide a good maintenance option, with close monitoring during and after steroid cessation. Having a relapse plan with readily accessible steroids for typical symptoms has also been used locally in some patients. In the event of a relapse, after acute treatment, prophylaxis with low-dose prednisone or monthly IVIg would be reasonable, with mycophenolate or rituximab as a next step. In the event of a patient having TM, more aggressive immunosuppressive therapy such as rituximab from an early stage would be indicated if the patient is not showing early improvement with steroids. Second-line agents such as mycophenolate may take months to reach their full efficacy, thus corticosteroids should not be rapidly weaned when adding on or switching therapeutic agents.
Limitations of this study include the retrospective design, the small number of patients in some treatment groups and the fact that some patients were on multiple therapeutic agents simultaneously. Some regression to the mean with ARRs cannot be excluded. Additionally, indication bias which is inherently present in observational studies, is acknowledged. Future studies with accumulated larger cohorts should identify clinical and radiological characteristics which may confound treatment assignment and combine them into a propensity score for use in multivariable models in order to mitigate some of the effects of indication bias. While we were able to perform multivariable logistic regression when comparing paediatric and adult patients in our cohort, due to relatively small numbers of patients in some of the treatment groups, we could not apply this when analysing our therapeutic data. Nevertheless, our current study systematically evaluates treatment responses in both children and adults in the largest reported cohort of relapsing MOG antibody-positive patients to date with an average of 5 years of follow-up, and provides important information on disease course, therapeutic efficacy and outcomes.
Given the emerging literature on the clinical and radiological phenotype and pattern of treatment response, we believe this condition warrants designation as a separate clinical entity from MS and NMOSD. We recommend the terminology ‘MOG antibody-associated demyelination’ as a more accurate syndromic diagnosis. Given the high pretest probability of MOG antibody-positivity in children, we believe that it is reasonable to test for MOG antibodies in all childhood-onset demyelination, particularly if relapsing. In adults, we recommend that MOG antibody-testing could be reasonably restricted to patients with a clinical and radiological phenotype atypical for MS, particularly in the event of isolated or recurrent ON. The clinical spectrum in adults additionally appears to be broader than simply AQP4 antibody-negative patients with NMOSD.
In conclusion, ON is the dominant manifestation of MOG antibody-associated demyelination, but there is a wide clinical spectrum. Relapsing patients are often steroid responsive, but vulnerable to relapses on tapering or withdrawal. Consideration should be given to early secondary prevention treatment strategies that may include maintenance corticosteroids, IVIg or immunosuppression with mycophenolate or rituximab.
Supplementary file 2
Supplementary file 3
Supplementary file 4
Supplementary file 9
![[SP9.jpg]](https://jnnp.bmj.com/content/jnnp/89/2/127/DC9/embed/inline-supplementary-material-9.jpg?download=true){kind=link}
Acknowledgments
The authors would like to thank all the patients and family members who participated in this study. The authors would like to thank Dr Maggie Wang for our use of the Flow Cytometry Core Facility of the Westmead Research Hub (Australia). The authors would also like to thank Ms Elizabeth Barnes for her assistance with statistical analyses.
References
Footnotes
FB and RCD contributed equally.
Contributors Study conception and design: SRa, FB, RCD. Sample acquisition: all authors. Acquisition of laboratory data: SRa, TKN, VM, FB. Acquisition of clinical data: SRa, RCD. Study supervision, data analysis and interpretation: SRa, RCD. Writing of first draft: SRa, RCD. Editing and revising final draft for intellectual content: all authors.
Funding This work was supported by the National Health and Medical Research Council (NHMRC) (Australia), the Petre Foundation (Australia), Multiple Sclerosis Research Australia (MSRA) (Australia), the National Blood Authority IVIg grant (Australia), the Brain Foundation and the Sydney Research Excellence Initiative 2020 Neuroimmunology Group (University of Sydney, Australia).
Competing interests Dr SR has received research funding from the National Health and Medical Research Council, the Petre Foundation and the Brain Foundation (Australia). Dr SM has received a scholarship from the National Health and Medical Research Council (Australia) and funding from the National Blood Authority IVIg grant. SB has received honoraria for attendance at advisory boards and travel sponsorship from Bayer-Schering, Biogen-Idec, Merck-Serono, Novartis and Sanofi-Genzyme, has received speakers honoraria from Biogen-Idec and Genzyme, is an investigator in clinical trials sponsored by Biogen-Idec, Novartis and Genzyme and was the recipient of an unencumbered research grant from Biogen-Idec. JL-S has accepted travel compensation from Novartis, Biogen and Merck-Serono. Her institution receives the honoraria for talks and advisory board commitment as well as research grants from Bayer Health Care, Biogen, Sanofi-Genzyme, Merck, Novartis and Teva. SR reports grants and personal fees from Sanofi-Genzyme, personal fees and departmental support from the Government of Australia, Baxter, Biogen, CSL and Merck; and departmental support from Novartis, outside the subject of the submitted work. Associate Professor FB has received research funding from the Star Scientific Foundation, The Trish Multiple Sclerosis Research Foundation, Multiple Sclerosis Research Australia and the National Health Medical Research Council (Australia). RD has received research funding from the Star Scientific Foundation, The Trish Multiple Sclerosis Research Foundation, Multiple Sclerosis Research Australia, the Petre Foundation and the National Health Medical Research Council (Australia). Dr RCD has received honoraria from Biogen-Idec as an invited speaker. Dr JLB has received compensation for education travel, honoraria for talks and advisory boards from Biogen, Teva, Merck-Serono, Sanofi-Genzyme, Novartis and Roche. Dr AC has received honoraria for attendance at advisory boards from Biogen and is an investigator in clinical trials sponsored by Biogen and Pfizer. Dr PC has accepted travel compensation from, Biogen and Merck Serono, and fellowship funding from Novartis/Biogen. Associate Professor CLF has received payment from Roche as a consultant. Dr MPM has received travel grants, speaking honoraria and unconditional research funding from Bayer, Biogen and Merck. Professor IES is supported by NHMRC Program Grant (1091593, 2016–2020) and Senior Practitioner Fellowship (1104831, 2016– 2020). IES serves on the editorial boards of Neurology and Epileptic Disorders; may accrue future revenue on a pending patent report: therapeutic compound; has received speakers honoraria from Athena Diagnostics, UCB, GSK, Eisai and Transgenomics; has received scientific advisory board honoraria from Nutricia and GSK, has received funding for travel from Athena Diagnostics, UCB and GSK and receives/has received research support from the NHMRC, ARC, NIH, Health Research Council of New Zealand, March of Dimes, the Weizmann Institute, CURE, US Department of Defense and the Perpetual Charitable Trustees. Dr EY is supported by an NHMRC Early Career Fellowship (APP1073323).
Ethics approval Sydney Children’s Network and Westmead Hospital Human Ethics Committees (12/SCHN/395, SSA/13/WMEAD/53).
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators Andrews PI (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia; School of Women’s and Children’s Health, University of New South Wales, Sydney, Australia), Barton JL (Brain and Mind Centre, University of Sydney, Sydney, Australia; Department of Neurology, Royal Prince Alfred Hospital, Sydney, Australia), Burrow JNC (Royal Darwin Hospital, Northern Territory Medical Program, Darwin, Australia; Flinders University of South Australia, Adelaide, Australia), Butzkueven H (Department of Medicine, University of Melbourne, Melbourne, Australia), Cairns AG (Children’s Health Queensland, South Brisbane, Australia), Calvert S (Children’s Health Queensland, South Brisbane, Australia; Department of Neurosciences, Lady Cilento Children’s Hospital, South Brisbane, Australia), Caruana P (Department of Neurology, John Hunter Hospital, Newcastle, Australia; Hunter Medical Research Institute, Faculty of Medicine and Public Health, University of Newcastle, Newcastle, Australia), Chelakkadan S (Department of Neurosciences, Lady Cilento Children’s Hospital, South Brisbane, Australia), Clark D (Women and Children’s Hospital, Adelaide, Australia), Fraser CL (Save Sight Institute, University of Sydney, Sydney, Australia), Freeman JL (Department of Neurology, Royal Children’s Hospital, Melbourne, Australia; Neurosciences Research, Murdoch Children’s Research Institute and University of Melbourne, Melbourne, Australia), Gill D, Grattan-Smith PJ (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia), Gupta S (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia; University of Sydney, Sydney, Australia), Hardy TA (Brain and Mind Centre, University of Sydney, Sydney, Australia; Department of Neurology, Concord Repatriation General Hospital, Sydney, Australia), Kothur K (Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead; Sydney Medical School, University of Sydney, Sydney, Australia; TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia), Ling SR (Neurology Service, Department of Paediatric Medicine, KK Women’s and Children’s Hospital, Singapore), Lopez JA (Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead; Sydney Medical School, University of Sydney, Sydney, Australia), Malone S (Department of Neurosciences, Lady Cilento Children’s Hospital, South Brisbane, Australia), Marriott MP (Department of Neurology, Royal Melbourne Hospital; Eastern Health Clinical School, Monash University, Melbourne, Australia), Nosadini M (Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead; Sydney Medical School, University of Sydney, Sydney, Australia; Paediatric Neurology and Neurophysiology Unit, Department of Women’s and Children’s Health, University Hospital of Padua, Italy), O’Grady GL (Paediatric Neuroservices, Starship Children’s Health, Auckland District Health Board, Auckland, New Zealand), Orr CF (Department of Neurology, Royal Perth Hospital, Perth, Australia), Ouvrier R (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia; University of Sydney, Sydney, Australia), Parratt J (University of Sydney, Sydney, Australia; Department of Neurology, Royal North Shore Hospital, Sydney, Australia), Patrick E (School of Mathematics and Statistics and the Westmead Millenium Institute For Medical Research; The University of Sydney, Sydney, Australia), Pilli D (Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead; Sydney Medical School, University of Sydney, Sydney, Australia), Riminton DS (Department of Neurology, Concord Repatriation General Hospital, Sydney, Australia), Riney K (Department of Neurosciences, Lady Cilento Children’s Hospital, South Brisbane, Australia; School of Medicine, University of Queensland, Brisbane, Australia), Rodriguez-Casero V (Department of Neurology, Royal Children’s Hospital, Melbourne, Australia; Monash Medical Centre, Melbourne, Australia), Ryan MM (Department of Neurology, Royal Children’s Hospital, Melbourne, Australia; Neurosciences Research, Murdoch Children’s Research Institute and University of Melbourne, Melbourne, Australia), Scheffer IE (University of Melbourne, Florey Institute, Austin Health, Melbourne, Australia), Shah UH (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia; Department of Neurosciences, Lady Cilento Children’s Hospital, South Brisbane, Australia), Shuey N (Neuro-ophthalmology clinic, Royal Victorian Eye and Ear Hospital and Department of Clinical Neurosciences, St Vincent’s Hospital, Melbourne, Australia), Spooner CG (Paediatric Neuroservices, Starship Children’s Health, Auckland District Health Board, Auckland, New Zealand), Subramanian GM (Department of Paediatrics, John Hunter Children’s Hospital, Newcastle, Australia), Tea F (Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead; Sydney Medical School, University of Sydney, Sydney, Australia), Thomas T (Neurology Service, Department of Paediatric Medicine, KK Women’s and Children’s Hospital, Singapore), Thompson J (Department of Neurology, St George Hospital, Sydney, Australia), Troedson C (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia), Ware TL (Department of Paediatrics, Royal Hobart Hospital, Hobart, Australia), Webster RI (TY Nelson Department of Neurology and Neurosurgery, Children’s Hospital at Westmead, Sydney, Australia; Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead, Sydney, Australia), Yiannikas C (University of Sydney, Sydney, Australia; Department of Neurology, Concord Repatriation General Hospital, Sydney, Australia; Department of Neurology, Royal North Shore Hospital, Sydney, Australia), Yiu EM (Department of Neurology, Royal Children’s Hospital, Melbourne, Australia; Neurosciences Research, Murdoch Children’s Research Institute and University of Melbourne, Melbourne, Australia), Zou A (Brain Autoimmunity Group, Institute for Neuroscience and Muscle Research, The Kids Research Institute at the Children’s Hospital at Westmead; Sydney Medical School, University of Sydney, Sydney, Australia).
Linked Articles
- Editorial commentary