Article Text

Abstract
Dystonia is a disorder of motor programmes controlling semiautomatic movements or postures, with clinical features such as sensory trick, which suggests sensorimotor mismatch as the basis. Dystonia was originally classified as a basal ganglia disease. It is now regarded as a ‘network’ disorder including the cerebellum, but the exact pathogenesis being unknown. Rare autopsy studies have found pathology both in the striatum and the cerebellum, and functional disorganisation was reported in the somatosensory cortex in patients. Recent animal studies showed physiologically tight disynaptic connections between the cerebellum and the striatum. We review clinical evidence in light of this new functional interaction between the cerebellum and basal ganglia, and put forward a hypothesis that dystonia is a basal ganglia disorder that can be induced by aberrant afferent inputs from the cerebellum.
- dystonia
- cerebellar disease
- functional imaging
- movement disorders
- neurophysiology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Dystonia is a syndrome of sustained muscle contractions frequently producing twisting, repetitive or patterned movements or abnormal postures.1 It often starts in a task-specific context.2 For instance, a patient with writer’s cramp, a focal dystonia, has difficulty in writing, but the performance of other elaborate tasks, such as using chopsticks, remains intact (online supplementary video: segments 1 and 2). These tasks are normally performed semiautomatically with little intentional effort of the subject. This characteristic implies a subconscious mechanism of activating a set of muscles specific for a particular automatic task (a motor subroutine).3
Supplementary video
Historically, this disease had been treated as a psychogenic or functional disorder,4 partly because of the clinical feature of sensory trick, in which the patient can benefit from particular cutaneous or proprioceptive sensory inputs usually unrelated to performing the task or maintaining the posture5 (figure 1). This trait, however, gave a clue to the understanding of its pathophysiology as a mismatch of sensory input versus motor output for the task or the posture.

A classical picture of sensory trick (geste antagoniste) in spasmodic torticollis.5 Left: without trick. Right: abnormal posture is corrected by placing the right hand on the hip bone and touching the chin with the left hand.
Is dystonia a sensory disorder?
Muscle afferents are the major inputs for kinesthetic sensation, and spindle afferent pathways project densely to the cerebellum via spinocerebellar tracts. Almost a century ago, Walshe6 reported that a diluted local anaesthetic can improve akinesia in a man with Parkinson’s disease, reasoning that this treatment altered muscle afferents. Diluted local anaesthetics block first nerve fibres with the smallest diameters. If injected diffusely into a muscle, they predominantly affect the gamma efferent fibres that innervate the spindles. Thus, this manoeuvre desensitises the spindles and secondarily blocks the muscle afferents (muscle afferent block or MAB).7
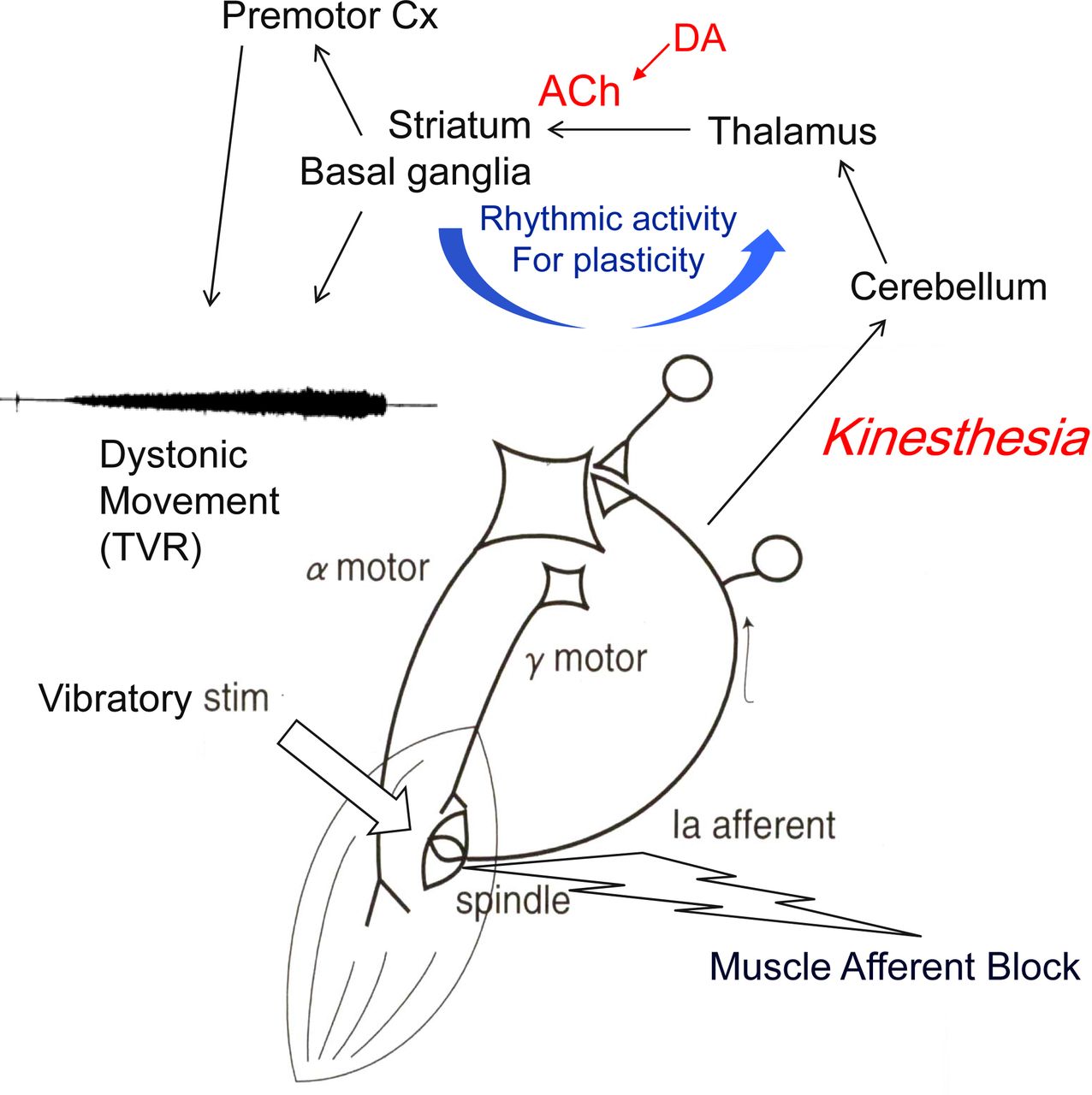
This effect of diluted lidocaine injected into dystonic muscles has also been explored, and MAB abated the abnormal contractions, although temporarily.8 The tonic vibration reflex (TVR) is a manoeuvre to activate muscle spindle afferents by applying high-frequency, large-amplitude vibratory stimulation to the belly of the muscle or to the associated tendon. If the TVR is tested for the limb affected by dystonia, it indeed reproduces dystonic contractions without the subject’s intention to perform the task with the affected limb (online supplementary video: segment 3). The exact neural pathways affected in the TVR are unknown, but the reflex is considered as subcortical, as cortical lesions do not abolish it.9 These findings suggest that dystonic movements or postures are already preprogrammed in a subcortical circuit even at rest, forming an abnormal sensory versus motor link (figure 2).
Subsequent functional studies focused attention to the disorganisation of the primary somatosensory cortex,10 raising much interest in the sensory aspects of dystonia.11 Further studies demonstrated abnormal temporal discrimination in the affected limb,12 impaired cortical inhibition of the surrounding muscles (surround inhibition13) and maladaptive cortical plasticity as shown by priming sensory stimuli and the associated motor excitability (paired associated stimulation),14 which are functional hallmarks of focal dystonias. It is not fully understood whether these cortical findings reflect primary or compensatory events in the genesis of dystonia.
Lessons learnt from X-linked recessive dystonia-parkinsonism and dopa-responsive dystonia (DRD)
X-linked dystonia-parkinsonism (XDP or DYT3) is a neurodegenerative disease affecting the basal ganglia and is endemic in the Panay Island in the Philippines. Its clinical features typically appear in the mid-30s as a focal dystonia followed within several years by parkinsonism.15 Autopsy studies in the early dystonic stage showed patchy lesions in the striatum, and those in the later parkinsonian phase exhibited total atrophy of the entire dorsal striatum similar to multiple systems atrophy.16 Detailed immunohistochemical studies have suggested that the striatal projection neurons (medium spiny neurons (MSNs)) affected in the early dystonic phase of the disease likely are located in striosomes, a neurochemically defined compartment in the striatum, as opposed to the striatal matrix, for which a marker (calbindin) remained (figure 2). No recognisable abnormalities were observed in the cerebellum in these cases.
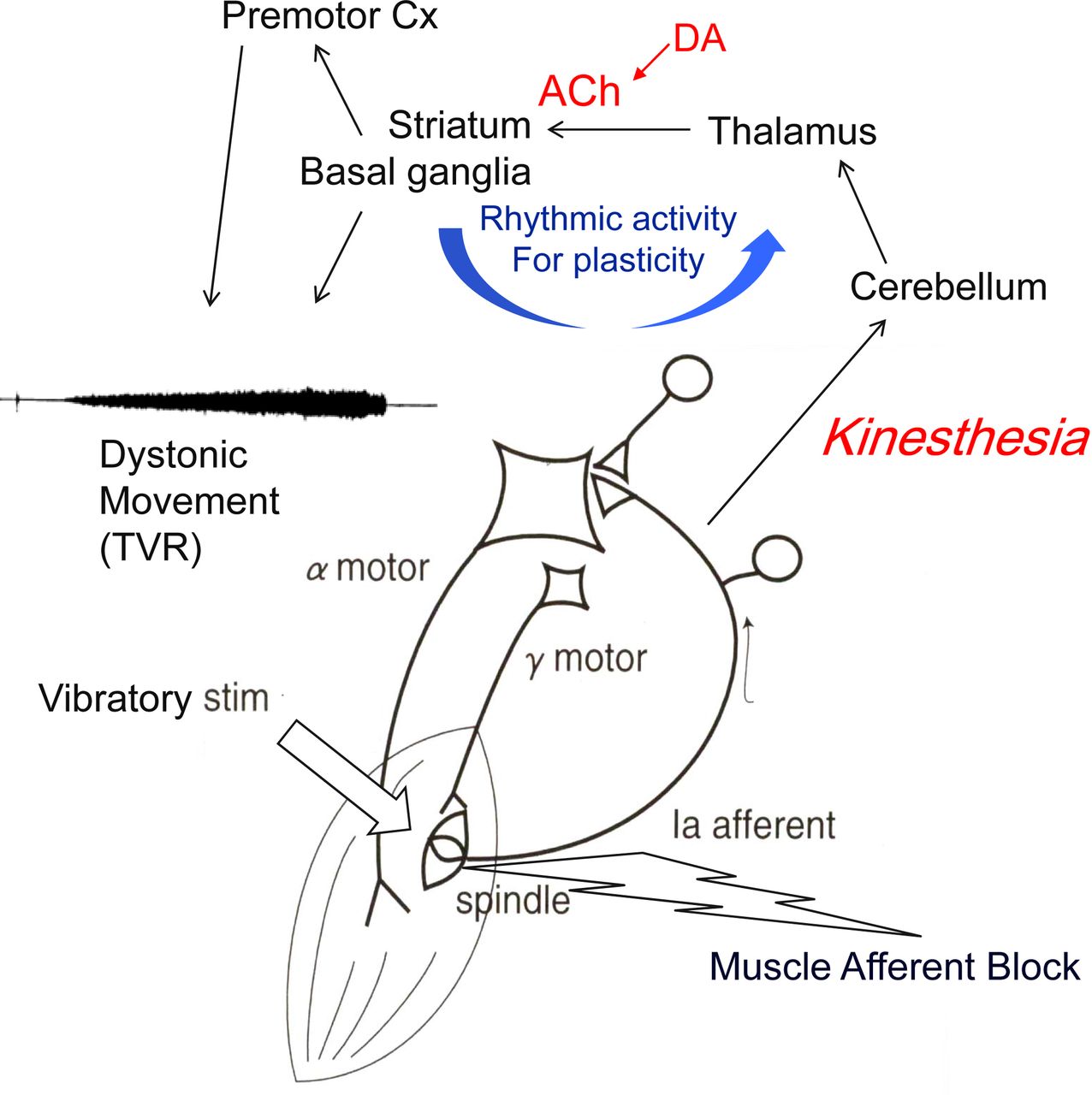
Scheme of hypothetical pathways for sensory versus motor output in dystonia. Dystonic movements are reproduced with high-frequency vibration (TVR) and blocked by desensitising the muscle spindle with diluted local anaesthetics (MAB). Spindle afferents are projected to the cerebellum, and disynaptic projection of cerebello-thalamo-striatal pathway affects the neuroplasticity at the corticostriatal synapse, where rhythmic activity is essential. Subcortical route is assumed for TVR. ACh, acethylcholine; DA, dopamine; MAB, muscle afferent block; TVR, tonic vibration reflex.
These findings drew attention to the three-compartment model of the basal ganglia circuit.17 The striosomes and the matrix are the major neurochemically defined compartments of the striatum. It is mainly the large matrix compartment that houses the neurons giving rise to the excitatory direct and the inhibitory indirect pathways that are standard components of the push–pull model of the basal ganglia.18 Given that dystonia is characterised by the activation of muscles unnecessary for the task (activation of antagonists or distant muscles; cocontraction or overflow), the direct pathway is proposed to be hyperactive.
The function of striosomes is not fully understood, but they receive inputs from regions in and connected with the limbic system.19 Their activity is influenced by local cholinergic interneurons, and they project directly to subsets of the dopamine-containing neurons of the substantia nigra pars compacta, thus placing them as important links in nigro-striato-nigral loops implicated in both neurological and neuropsychiatric disorders.20 The relationships among direct, indirect and striosomal pathways are currently a subject of great interest, particularly in light of recent evidence for the reciprocal interactions of the striatum with its dopaminergic afferents (figure 3).21 There may be collateral influences across striosome and matrix compartments involving the dopaminergic afferents,22 suggesting that, along with cholinergic crosstalk between the compartments, there may be dopaminergic crosstalk as well. One possibility is that the nigro-striosomal circuit is suited for feedback inhibitory control of dopamine release in the striatum and even within the substantia nigra itself. If so, the preferential loss of striosomes reported to occur in XDP could lead to disinhibition of dopamine release with relative direct pathway predominance, which could account for dystonia (figure 2).

{kind=link}
{kind=link}
{kind=link}
Striosomes (S) and direct versus indirect pathways in the matrix (M) in normal (left) and XDP dystonia (right). The lower left subset indicates activated (red) and inhibited (blue) muscles for the task (surround inhibition). The loss of striosomal neurons in XDP disinhibits dopaminergic release by substantia nigra pars compacta (SNc, dotted arrow), which in turn makes direct pathway predominant over indirect pathway, resulting in disruption of surround inhibition. GABA; gamma-aminobutyric acid; XDP, X-linked dystonia-parkinsonism.
DRD23 or Segawa disease (DYT5) seems contradictory to a theory of dystonia based on an overabundance of dopamine, because DRD readily responds to l-dopa treatment. Sato and colleagues24 resolved this issue by examining the striosome and matrix compartments immunohistochemically in a mouse model of DRD. Using tyrosine hydroxylase (TH) staining, they found early selective loss of TH immunostaining in striosomes, suggesting that a lack of nigrostriatal dopamine D1 input to the striosomal MSNs could produce deficient GABAergic inhibition of neurons in the substantia nigra pars compacta with relative direct pathway predominance in the matrix, where TH activity remains intact. A single autopsy report of patient with DYT5 also showed striosomal decrease of TH staining, lending support to this hypothesis.25
Is dystonia a cerebellar disorder?
It has been reported that dystonia could be the presenting symptom in the spinocerebellar ataxias (SCA2, 3, 6 and 17). Among these, SCA6 is associated with pathology confined to the cerebellum. Furthermore, cerebellar atrophy or signs have been noted in late-onset dystonia.26 A family with marked cerebellar atrophy presenting predominant dystonia has also been described.27 Miyamoto and colleagues28 reported an autopsy study of such a family. The patient presented mostly with generalised dystonia that partially responded to deep brain stimulation (DBS) of the globus pallidus internus (GPi). The pathological finding was pure cerebello-olivary degeneration with no abnormalities in the basal ganglia. The fact that DBS was effective indicates that the basal ganglia dysfunction, if any, is downstream effects of the cerebellar lesion.
Animal studies also provided clues to the cerebellar involvement in dystonia. DYT1 model mice exhibit increased energy metabolism in the olivocerebellar network.29 Phenotypes of pharmacologically or genetically modified animal models of cerebellar origin are markedly aggravated by the dysfunction of the basal ganglia.30 Conversely, dystonic features are improved by lesions of the thalamic relay nuclei linking the cerebellum and the striatum and are eliminated in another genetic model in the rat by cerebellotomy.31 These findings indicate that aberrant inputs from the cerebellum to the basal ganglia may underlie the genesis of dystonia.
Despite these findings favouring a cerebellar origin for dystonia, another recent review32 concluded that ‘the cerebellum plays a role in the pathophysiology of dystonia, but do not provide conclusive evidence that the cerebellum is the primary or sole neuroanatomical site of origin’. Supporting pieces of evidence of this view include the lack of case reports of focal lesions in the cerebellum in focal dystonias despite many reports of focal lesions in the basal ganglia in such dystonias.33 34 DBS is successful in treating focal dystonia when targeted at the GPi or ventral oral thalamus, which are parts of the basal ganglia motor loop, but not, as currently recognised, in the cerebellar recipient thalamus (ventral intermediate).35
Is dystonia of cerebral cortical origin?
Using positron emission tomography and diffusion tensor imaging (DTI) to judge white matter integrity, Carbon and colleagues,36 in the patients with DYT1 and DYT6 genotype, found phenotype-related and genotype-related brain activation patterns, focusing on the basal ganglia (cortico-striato-pallido-thalamo-cortical) motor loop and related cerebellar (cerebello-thalamo-cortical) circuits. The phenotype-related pattern, which could include compensatory mechanisms secondary to the genotype, was associated with increases in the presupplementary motor area and parietal association areas of the cerebral cortex, but the genotype-specific or intrinsic abnormalities were localised to the cerebellum, basal ganglia and their output cortical region in the supplementary motor area. More recently, the same group37 reported DTI findings on the cortical white matter underlying the affected and unaffected body part representations in dystonia. They found that white matter integrity was reduced in regions related to the unaffected body parts but was normal in those related to the affected parts, indicating that reduced integrity in the unaffected subcortical region is secondary. These findings suggest that the intrinsic abnormality in dystonia is likely at the subcortical level of the basal ganglia and the cerebellum and that the cortical events are downstream compensatory effects.
A new pathway that enables direct interaction between the cerebellum and the striatum
The cerebellum and the basal ganglia have been thought to communicate with each other only through multisynaptic, principally cortical circuits. Anatomical tracing studies in rodents and primates have now shown, however, a direct disynaptic projection from the cerebellar nuclei to the basal ganglia via the intralaminar thalamic nuclei.38 It has also been found that an intralaminar thalamo-striatal projection can trigger striatal dopamine release by engaging the synchronised activity in cholinergic interneurons that synapse on the terminal arborisations of dopaminergic projections to MSNs.39
Recently, Chen and colleagues40 have further explored this disynaptic short-latency (~10 ms) projection from the cerebellum to the striatum using electrophysiological recordings in awake mice. They found that the cerebellum rapidly modulates the activity of the striatum via this disynaptic pathway. In their studies, high-frequency stimulation of the neocortex alone predominantly depressed corticostriatal responses (long-term depression), whereas concurrent stimulation of the cerebellum altered the plasticity towards enhanced synaptic efficacy (long-term potentiation (LTP)). In a previous study of a mouse model of DYT1,41 depotentiation of corticostriatal LTP became no longer possible, suggesting that abnormal synaptic plasticity is the underlying mechanistic dysfunction in dystonia. In their model of rapid-onset dystonia-parkinsonism, Chen and colleagues further reproduced burst-like activity in striatal neurons by selective partial block of a cerebellar sodium pump. These dysrhythmic activities are similar to those seen in the GPi at the time of DBS in human dystonia. They also found that cerebellar-induced dystonia was alleviated by severing the link between the cerebellum and the basal ganglia, indicating that aberrant cerebellar inputs could cause dystonia.
The mechanism by which DBS improves dystonia is still unknown. The burst-like activities of the basal ganglia neurons, which can be produced by aberrant cerebellar input, may interfere with the control of synaptic plasticity by frequency modulation, and high-frequency overdriving of the basal ganglia circuits by DBS possibly rescues the abnormal neuroplasticity in dystonia. It is of note that anticholinergics also restore the impaired synaptic depotentiation of LTP seen in DYT1 model, possibly because of the link of cholinergic and dopaminergic projections converging at the striatum.42
Revised view of the network affected in dystonia
Dystonia can be regarded as a disorder of aberrant input versus output plasticity or sensorimotor mismatch, which is clinically exemplified by ‘sensory tricks’ or other sensory phenomena. Dystonic contractions can be reproduced by stimulating muscle spindle afferents with TVR even in task-specific cases (video) and are abolished after blocking muscle afferents. The pathway for this reflex remains unknown, but it could use the cerebellum as the afferent limb because spindle afferents are not perceived at the conscious level, and they have the dense forward projections to the cerebellum. The TVR persistence after cortical lesions in patients with stroke also suggests its subcortical routes. Finally, TVRs are greatly attenuated by cerebellar lesions.43
Dystonia has traditionally been regarded as a basal ganglia disorder.44 The relative lack of dopamine in striosomes could produce DRD by deranging its dopamine sensor function in the striatum. As discussed, a pathological finding was documented only in the striosomes at the early dystonic phase of XDP. The discovery of the functional disynaptic short-latency pathway from the cerebellum to the basal ganglia raises the possibility that cerebellar dysfunction could also cause dystonia by sending aberrant input to the striatum, where abnormal control of neural plasticity occurs as a result. Questions still remain pertaining to the role of cholinergic fibres in these circuits, the mechanisms by which striosomes function and how the cerebellum sends its sensory feedback to the striatum. The relation of rhythmic activity in the basal ganglia to the clinical effect of DBS is a further issue to be clarified.
Conclusion
Clinical features of dystonia underscore the sensory aspects of its symptoms, and indeed the premise that dystonia is a sensory disorder appears to be correct. If the afferent or sensory limb of the motor coordination of semiautomatic tasks involved in dystonia uses the spinocerebellar and the cerebello-thalamo-striatal pathway, it resolves the enigma of the cerebellar origin of dystonia.
Acknowledgments
We would like to thank S Goto in Tokushima University for their helpful advice and Y Kubota in MIT for their kind editing.
References
Footnotes
Contributors RK wrote the first draft, and KB and AMG reviewed and revised it.
Funding Funded by grants from Japanese Ministry of Health, Welfare and Labor (H28-019) and the Collaborative Center for X-linked Dystonia Parkinsonism at Massachusetts General Hospital.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed.