Article Text

Statistics from Altmetric.com
Introduction
The adult neurodegenerative disorder amyotrophic lateral sclerosis (ALS) is unmistakeable from clinical descriptions now more than a century old.1 Neither the site of symptom onset in ALS2 nor pattern of symptom spread3–5 is truly random. While the most consistent signs relate to motor system dysfunction, it is recognised that ALS represents an anatomically widespread neocortical disease,6 involving significant pathological and genetic overlap with Frontotemporal Dementia (FTD).7 8 Nearly all cases of ALS, and half of FTD, are associated with cytoplasmic, ubiquitinated inclusions of aggregated TDP-43.9 The linkage of apparently disparate cortical networks in the ALS-FTD pathological spectrum has been conceptualised as ‘what wires together, fires together and dies together’,10 based on a wider concept that neurodegenerative syndromes are defined by the architecture of large-scale brain networks.11 The motor system is perhaps one of the most fundamental to evolution, and the relative rarity of its targeted disintegration within the spectrum of neocortical disorders12 encapsulates a fundamental conundrum of understanding selective vulnerability in cerebral neurodegeneration.13
Approximately 600 000 years ago, Homo sapiens evolved from a common ancestor along with Neanderthal and other Hominid species, with evidence that Homo Sapiens of African origin exhibited the greatest genomic diversity.14–16 During this time, efficient cerebral processing for complex functioning evolved. This included versatile vocalisation accompanied by complexity of respiration, language and associated socialisation, increased fractionation of digits and thumb opposability, and upright walking with ability to navigate uneven and tricky surfaces while erect (figure 1). Failure of all of these functions characterises clinical manifestations of ALS.17 Paralleling functional advances, the shape and development of the brain have also evolved,18 including a diversification of different cell types. To achieve the fast, efficient motor processing required, particular motor output systems developed, best exemplified by the monosynaptic corticomotoneuronal system. Efficient processing was further enhanced by increased cerebral dominance, where laterality could maximise efficiency. We now consider the evolutionary biology of hemispheric specialisation in more depth as it might pertain to selective vulnerability in ALS.
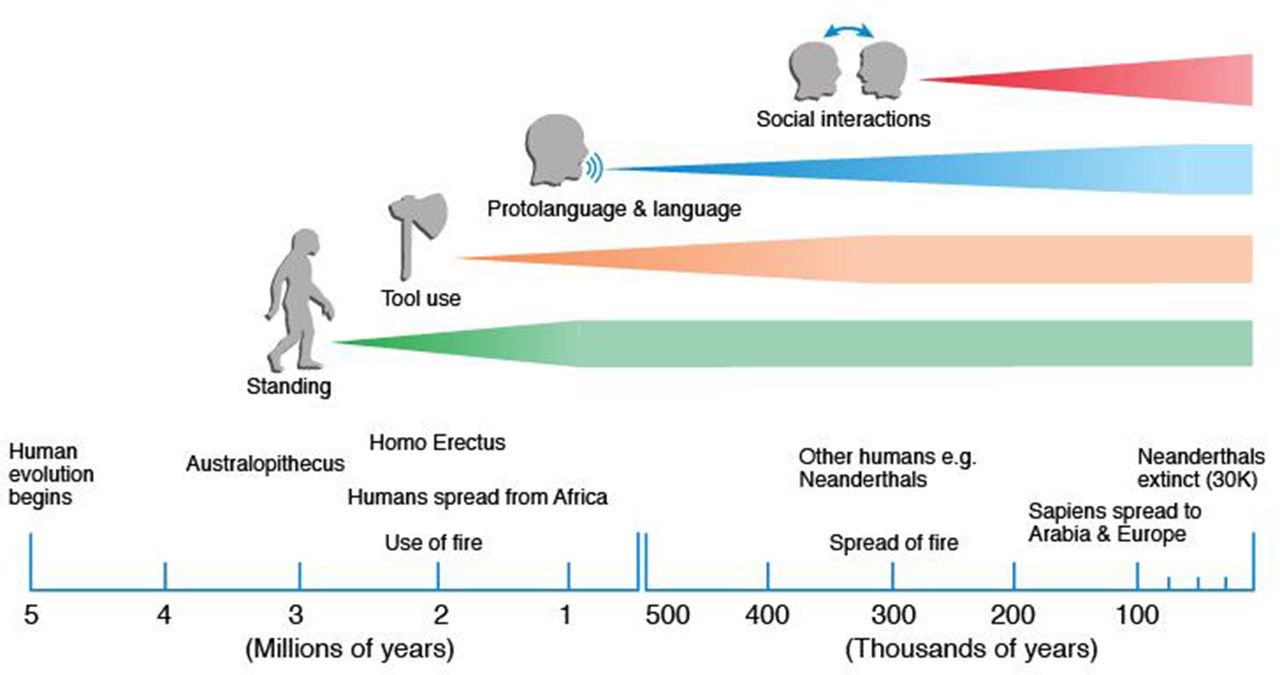
A timeline of human development within the concept that this is very approximate and likely to change with new information which typically causes the lines to move to the left. The definitions of use of tools and social interaction are not defined. Standing and use of tools occurred across different humans (eg, including Neanderthals) but language and social interaction in the last 70 000 years is only clear for Homo sapiens.
Hemispheric dominance
Given that there is widespread cerebral dominance in vertebrate brains, and even some invertebrates, it seems reasonable to assume that this must confer favourable selective advantages,19 20 possibly by increasing neural capacity, within the limitations of a bony skull.21 Specialisation within each hemisphere enables the brain to control specific functions such as handedness, gesturing and speech, but also vision and other functions such as sleep.22 Humans are uniquely predominantly right-handed, and most right-handed individuals are left hemisphere dominant for language. Typically, 97% of right-handers demonstrate left-brain preference for language but so too do 60% of left-handers.23 24
Cerebral dominance is limited in non-primates, becoming increasingly evident with primate evolution, and most apparent in humans.23 Nevertheless, left-right asymmetries are found in many animal species (action dynamics for aquatic mammals, vocalisation for mice and amphibians).25 Humans however have the most consistent population-wide bias towards cerebral laterality, and impaired dominance is associated with a number of disorders (such as autism and perhaps schizophrenia).26–28
Left hemispheric preference in primate development possibly occurred as a random event, although evidence favours a link with language dominance, in that language dominance and handedness were considered to have co-evolved in hominid evolution.25 29 However, contrary evidence from behavioural and neuroimaging studies challenges this and biological, functional and social factors have also been shown to be determinants of hand dominance.30 For example, hand preferences in non-human primates reveal evidence of population-level handedness, but during some tasks, requiring controlled posture and reaching, the majority of prosimians show left-handedness.31 In Old and New World monkeys as well as great apes, coordinated bimanual tasks show consistent evidence of population-level handedness. In rhesus monkeys and chimpanzees, there is preferential use of the right hand for gesturing, and this is enhanced when accompanied by a vocalisation.32 While other species have varying degrees of dominance, humans have the most consistent population-wide bias toward right upper limb dominance.25 This preference in modern man for right-handedness possibly dates to at least 500 000 years,33 perhaps to a linkage with the use of tools (figure 1).
Two theories have developed to explain specialisation of the left hemisphere for language, handedness and other functions.32 One advocates that specialisation is primarily genetically determined becoming evident at an early developmental stage, predisposing one hemisphere to ‘take the lead’.34 An alternative model promotes the corpus callosum as key during prenatal and postnatal development enhancing cross-hemispheric communication.35 Neuronal influences are mediated through the fibres of the corpus callosum, which arise from cortical pyramidal cells that are largely glutamatergic, affecting an excitatory role, although they are also known to act through gamma aminobutyric acid-mediated inhibitory neurons.36 At a functional level, the corpus callosum enhances processing across the cerebral hemispheres (excitation theory).37 Alternatively, its primary function could be transmission of information across cortical fields to suppress one area while the contralateral hemisphere is active.38 There is a possible trade-off between relative corpus callosum size and the speed and architecture of the fibres traversing the corpus callosum.32
Cerebral dominance can also be considered within global cerebral connectivity concepts where there are favoured resting state networks that facilitate pathways of common use.39 40 Nodal signalling and other pathways appear to have a genetic basis,21 suggesting that these pathways developed over a long evolutionary period of time, across many animal species,21 and ultimately for specific human brain processing functions such as right-hand dominance, language and socialisation.41
As the human brain adapted to accommodate more complex motor and social skills, there was a need for fast, efficient cerebral processing within the cerebral cortex and particularly the frontal lobes, which expanded disproportionately to other brain regions.42 43 This was accompanied by extensive structural changes, including globularisation,18 neural pruning, myelination and synaptogenesis, proceeding from the first year of life through to early teenage years.29 44 At the same time, complex neural networks disproportionately evolved over a relatively shorter time span, enabling specific human functions.45 Such developments potentially rendered the neocortex, in particular more vulnerable to neurodegeneration.12
An illustration of one such possible association of the concepts of lateralised circuits and evolution could be the evolutionary use of fire. Humans obtained an ability to use fire relatively late in evolutionary terms. This required use of tools to ‘make it and keep it’, with the development of handedness becoming a distinct advantage. Further, social interaction occurred as people gathered for the benefits of fire including warmth, cooking, photoperiodicity and the ability to disperse to colder climates,46 accelerating the development of language which was needed as people interacted. All of these cerebral modifications developed over a relatively rapid period with vulnerability of neurons to the effects of ageing.
The ALS-FTD syndrome as a disorder of vulnerable cerebral networks
There is increasing appreciation that ALS and FTD are disorders of impaired cerebral network connectivity.47 ALS can be considered, at its clinical core, to be a progressive disruption of the complex connection between the corticomotoneuron and targets muscles that implement specific adaptive, complex functions.48 Failure of the corticomotoneuronal system preferentially involves the complex adaptive motor skills that are most highly developed in modern humans, such as control of the thumb and index finger, upright walking and speech.17 49 The focal nature of ALS is exemplified, by the preferential involvement of the lateral portion of the hand, termed the ‘split hand syndrome’.50 51 There is greater cortical representation and connectivity of the thenar hand14 and current evidence favours a disease mechanism occurring at a cortical level.52–54 The focal loss of functionality of the thumb in ALS as a reflection of evolutionary motor system specialisation was enhanced by the recognition that speech (a later evolutionary development) is involved before swallow in ALS, particularly in those with upper motor neuron pathology,5 55 and a preferential involvement of gait,54 language56 and social awareness across the ALS-FTD spectrum.17
Although overt clinical deficit is likely to be a late manifestation of ALS pathology,57 once apparent, symptoms inexorably progress in the region where the disease commenced. In some patients, it remains initially restricted to that region for an extended period of time, while in others it appears to have a multifocal and rapid progression.58 In ALS, the relatively long preservation of eye movements and sphincter function, both innervated through indirect, polysynaptic cortical pathways, which are in evolutionary terms, old and conserved, as opposed to direct corticomotoneuronal connections which are of recent origin and therefore more vulnerable, would further support evolutionary concepts.48
The non-motor cerebral regions may show some of the greatest atrophy when the disease is well established.59 As a case in point, an ALS case where the disease process continued while the patient was mechanically ventilated in a persistent vegetative state for 8 years illustrated the striking pathological involvement mainly of the frontal lobe regions (figure 2). Atrophy that occurs in these frontal areas is morphologically opposed to the cortical development that has developed in these regions through evolution from the primate.

Striking atrophy of frontal lobe regions in a patient with ALS who remained in a persistent vegetative state for 7 years. The clinical presentation was typical of ALS but cognitive studies were not performed. Similarities of the loss of frontal lobe regions with the limited development of these regions in primates and possibly early humans is depicted in the right-hand panel. ALS, amyotrophic lateral sclerosis.
Applying lateralisation of cerebral functions to ALS
In ALS, cerebral dominance in relation to handedness (though not necessarily foot preference) appears to influence clinical presentation2 and progression,5 so that upper limb onset ALS is more likely to occur in the dominant hand. Spread of weakness beyond the limb of onset also correlated with cerebral dominance, with onset in the non-dominant side more likely to progress to the ipsilateral non-dominant limb in comparison with the onset on the dominant side which spreads to the other contralateral side at that level.5 58 Similar patterns of contiguous spread have been noted in those with lower limb onset.4 These suggest a potentially important role for central (upper motor neuron) connections in influencing disease asymmetry. Supporting this theory, neuroimaging has demonstrated disproportionate atrophy of relevant cortical regions in patients with ALS with onset in the dominant hand60 (figure 3).

{kind=link}
{kind=link}
{kind=link}
Normally there is cortical asymmetry with a slightly larger and more distinct region in the left hemisphere (top panel). The solid arrow indicates cortical thickness. Loss of this cortical asymmetry occurs in patients with MND (bottom panel) in comparison with controls with preferential involvement (atrophy) of the dominant left hemisphere (arrow).60 MND; Motor Neurone Disease.
Cerebral dominance (a normal evolutionary process) and pathological processes can both be considered as a brain network connectivity modification, with the corpus callosum as an important determinant.32 Degeneration of the corpus callosum is a consistent finding in ALS pathology and is most salient within motor-associated fibre regions.61–63 Postmortem diffusion imaging has enabled correlation of microstructural imaging changes with the corresponding histological changes of axonal loss, astrocytosis and microglial infiltration,64 most prominent in patients carrying a hexanucleotide expansion in C9orf72, the most common hereditary form of ALS and behavioural variant FTD.
Local and regional cortical communication is associated with continuous rhythmic neuronal oscillations, which during movement preparation and execution, modulate at an oscillation of 15–30 Hz (beta), as measured by magnetoencephalography. Augmented beta desynchronisation in both ipsilateral and contralateral motor cortices has been described in patients with ALS during motor preparation.65 Further, in symptomatic carriers of genetic mutations, excess beta desynchronisation was recorded with movement execution. Movement completion was followed by a slowed rebound of beta power, postulated to correspond to involvement of interhemispheric fibres of the corpus callosum. Intensified cortical beta desynchronisation followed by delayed rebound is concordant with the broader concept of cortical hyperexcitability in ALS.66 Hyperexcitability can be hypothesised to occur to a greater degree in the dominant hemisphere, which has had, over a number of decades, greater functionality.
The evolutionary concepts pertaining to vulnerable cerebral circuits can be practically applied to the unanswered clinical observations in ALS such as the age of onset, gender preference and the lack of occurrence in non-human species or with other diseases. Primates and early humans, who had an expected lifespan of 40–60 years67 (ie, a similar to the typical mean age of onset for ALS), evolved lateralised pathways and enhanced cerebral connectivity suited to this lifespan. A clear difference in lateralised circuits is recognised between human genders and can be extended to include primates68 and might have relevance for the known gender inequality in ALS.69 The lack of occurrence of ALS pathology in non-humans and the relative lack of association of ALS with other organ diseases also become somewhat clearer if the evolution of lateralised cerebral circuits is considered as they pertain to humans.
Genetic considerations
Approximately one quarter of the variation in hand preference can be explained by additive genetic effects,70 however, these cannot be assumed to completely reflect cerebral dominance, as the traits are imperfectly related. Brain structure and symmetry correlate with hand preference and speech processing and have higher estimates of variation explained by genetic effects (33%–53%).71 These correlations imply cerebral dominance is a complex trait, underpinned by polygenic and non-genetic components. The genetic contribution to ALS pathogenesis is also considered complex, with both rare and common variation72–74 suggested to be part of a multistep process.75 For a carrier of a rare, high-penetrant disease-causing mutation, other factor/s still need to occur or accumulate over time to result in disease.
The presence and frequency of variants in the human genome can be influenced by the demographic histories of populations (ie, migration and natural selection).76 Most traits are polygenic and selection can be acting on many trait-associated variants simultaneously each with different effects on related phenotypes.77 Measuring genetic conservation in humans may provide a mechanism to understand links between evolution and function. Traditionally, this approach has been relatively limited as the human genome, in evolutionary terms, is relatively young. The amount of time for variants to accumulate for diversity (~200 000 years) and the size of the population (~10 000 breeding individuals) has resulted in a relatively small number of evolutionary differences between individuals and species.78 Put into perspective, with short-read technologies, the absolute sequence difference between chimpanzee and human is estimated to be ~4% of the genome, which includes coding differences (within the exome) of ~1% and non-coding changes of ~3%.79 80
Approximately 10% of ALS cases81 have been identified to carry rare coding variants in ~25 genes. Using available amino acid substitution quotients between human and chimpanzees80 and testing for enrichment for loss of function or non-synonymous intolerant genes in an exome population database82 do not support a hypothesis that the current known ALS genes have been under any positive selection pressure within or across species. Common single-nucleotide polymorphisms are also estimated to have a role in ALS pathogenesis,72 73 and while those identified explain <0.5% of the heritability, it is hypothesised that associated alleles produce a sub-threshold abnormality that in turn can be inherited.83 This suggests a model of ‘ancestral neutrality’.
Interestingly, common genetic variants associated with schizophrenia have found to have a higher propensity to be in genomic regions that diverge from their Neanderthal counterparts, than expected by chance.84 Such findings suggest that variants are associated with becoming human.85 If so, it could be hypothesised that other cerebral diseases developed as a result of genetic specialisation for functions specific to humans86and would have a higher frequency of occurring together.87 An alternative theory to ‘ancestral neutrality’, which has been proposed for variants associated with psychiatric and developmental disorders such as schizophrenia,84 85 is that there is both a negative and positive fitness effect that may explain why certain disease variants have been maintained in the population (ie, a balanced selection model). For common variants associated with a disease, evidence of a founder effect may indicate a positive effect that maintains the variant in the population. Multiethnic ALS population studies demonstrate relatively few founder effects (single ancestor) and the majority of ALS variants are rare and arise independently in a common set of known genes (ie, SOD1).88 89 An exception to this is the C9orf72 expansion,90 91 which does suggest a one-off expansion originating in Northern Europe ~6300 years ago.92 A balanced-selection model may suggest that this variant has not decreased in frequency due to a positive selection effect on cerebral connectivity. This is not dissimilar to hypotheses of ‘creative cognition’ whereby human evolution may have favoured this trait, intriguingly however it also may present genetic vulnerability for neurological and psychiatric disorders.93
The cellular processes that maintain the normal physiology of the neuronal system throughout life are currently being elucidated. Common genes identified in ALS and FTD function in a number of common molecular pathways involving RNA metabolism and homeostasis.94 95 In particular, the cellular accumulation of the DNA/RNA-binding protein TDP-43 (encoded by TARDBP) found in 90% of ALS cases highlights the importance of DNA/RNA homeostasis in neurons. Following DNA transcription, RNA molecules within a cell are bound by distinct sets of RNA-binding proteins that have the task of regulating the correct processing, transport, stability and function/translation up to its final degradation. Increasing age is the main risk factor for accumulation of protein aggregates as seen in neurodegenerative diseases with different protein signalling pathways affected depending on the unfolded protein response. As human lifespan has increased through evolution, there is a theory that cerebral protein aggregation has acted as a constraint on increasing age.96
Concluding remarks
We propose that ALS may be a disease that preferentially affects cerebral structures and pathways that have evolved recently in human evolution. Specifically, it is proposed that ALS-FTD represents a syndrome preferentially involving regions of the brain under relatively recent evolutionary change, whose complexity possibly renders it more vulnerable to insults or stressors that accumulate with ageing. The connectivity pathways serving rapid cerebral processing are sadly the most vulnerable to changes with increasing age.
Neurodegenerative syndromes have a complex polygenic basis resulting in impairment of neocortical network connectivity. Some profound psychiatric disorders of brain connectivity become clinically overt in much younger adults, but have a greater than expected incidence in those with ALS and their relatives,97 98 suggesting that neurodevelopment may have important implications for understanding neurodegeneration.
Motor system neurodegeneration, previously neglected, can be applied to the increasing literature relating to evolution and cerebral connectivity that has so far been dominated by language and wider cognitive processing considerations. Such concepts, while they are necessarily speculative, support the classification of ALS among the cerebral neurodegenerative rather than peripheral nerve disorders.
Acknowledgments
Naomi Wray, Matthew Devine and Roger Lemon provided valuable commentary during the evolution of the manuscript.
References
Footnotes
Contributors RH and AE: study concept and design. All authors: drafting of the manuscript. RH, AE, MK and MT: critical revision of the manuscript for important intellectual content. RH, FG and MK: administrative, technical or material support.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Correction notice Since this article was first published online there have been some text changes in the second paragraph of the introduction.