Article Text

Abstract
Objective A hallmark of amyotrophic lateral sclerosis (ALS) caused by mutations in superoxide dismutase-1 (SOD1) are inclusions containing SOD1 in motor neurons. Here, we searched for SOD1-positive inclusions in 29 patients carrying ALS-linked mutations in six other genes.
Methods A panel of antibodies that specifically recognise misfolded SOD1 species were used for immunohistochemical investigations of autopsy tissue.
Results The 18 patients with hexanucleotide-repeat-expansions in C9orf72 had inclusions of misfolded wild type (WT) SOD1WT in spinal motor neurons. Similar inclusions were occasionally observed in medulla oblongata and in the motor cortex and frontal lobe. Patients with mutations in FUS, KIF5A, NEK1, ALSIN or VAPB, carried similar SOD1WT inclusions. Minute amounts of misSOD1WT inclusions were detected in 2 of 20 patients deceased from non-neurological causes and in 4 of 10 patients with other neurodegenerative diseases. Comparison was made with 17 patients with 9 different SOD1 mutations. Morphologically, the inclusions in patients with mutations in C9orf72HRE, FUS, KIF5A, NEK1, VAPB and ALSIN resembled inclusions in patients carrying the wildtype-like SOD1D90A mutation, whereas patients carrying unstable SOD1 mutations (A4V, V5M, D76Y, D83G, D101G, G114A, G127X, L144F) had larger skein-like SOD1-positive inclusions.
Conclusions and relevance Abundant inclusions containing misfolded SOD1WT are found in spinal and cortical motor neurons in patients carrying mutations in six ALS-causing genes other than SOD1. This suggests that misfolding of SOD1WT can be part of a common downstream event that may be pathogenic. The new anti-SOD1 therapeutics in development may have applications for a broader range of patients.
- amyotrophic lateral sclerosis
- neuronal inclusions
- C9orf72
- KIF5A
- superoxide dismutase-1
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative syndrome characterised by adult-onset progressive loss of motor neurons with a focal onset of progressive paresis and muscle wasting.1–3
Some 5%–10% of patients with ALS self-report a familial predisposition (fALS), and the remaining cases are denoted as sporadic ALS (sALS). Since 1993, mutations in 37 genes have been reported to predispose to ALS. In Caucasians, the most commonly reported mutation is a GGGGCC repeat-expansion in C9orf72 (C9orf72HRE), found in 8%–10% of patients with ALS. Missense, substitution or deletion mutations in the genes encoding superoxide dismutase-1 (SOD1), TAR-DNA-binding protein 43 (TDP-43), fused in sarcoma (FUS) and kinesin heavy chain isoform 5A (KIF5A) are found in 2%–6% of European patients. The remaining gene mutations are rare. ALS is both genetically and clinically heterogeneous and the reported predisposing genes pleiotropic.4 Neuropsychological examinations and imaging studies can show involvement of the frontotemporal lobes,5 6 though this may remain subclinical.7 In particular, carriers of C9orf72HRE, FUS, VCP and TBK1 mutations may develop frontotemporal dementia (FTD) sometimes without overt ALS. Unfortunately, there have been few comparative neuropathological examinations in patients with different gene defects.
The first ALS-causing gene discovered was SOD1 8 and since 1993 more than 210 mutations have been found (http://alsod.iop.kcl.ac.uk/).9 While some of the mutant SOD1s (eg, C6S, D90A, E100K, L117V) are sufficiently stable and active to yield normal SOD1 activity in human tissues,10 others, for example, A4V, V5M, D76Y, D83G, G127Gfs*5 (alias ‘G127X’) are unstable, are rapidly targeted for degradation and can only be detected in minute amounts.11 12
Neuronal inclusions containing aggregated SOD1 are recognised pathological hallmarks of ALS caused by SOD1 mutations in patients and in transgenic (Tg) animal models overexpressing mutant human (h) SOD1s.11 13 In the Tg models, two structurally different strains have been found.14 When inoculated into the lumbar spinal cord of adult mice, both strains induce cell-to-cell propagating templated hSOD1 aggregation primarily in the motor neurons resulting in rapidly progressing ALS-like fatal diseases.15 Similar effects have been obtained inoculating SOD1-aggregates purified from the spinal cord of a patient with the SOD1G127X mutation.16 This prion-like action could be the primary pathogenic mechanism of SOD1-provoked ALS, which prompts exploration of SOD1 in ALS caused by other genes.
Previously, we examined spinal cord tissue from 37 patients with sALS and found numerous inclusions containing misfolded wild-type SOD1 (misSOD1WT) in the cytoplasm and nuclei of neurons and glial cells.17 18 The presence of misSOD1WT in motor neurons of patients with ALS was later observed by other groups with the differences that antibodies against other SOD1-epitopes were used and inclusions were not found in all patients studied.19–22 Supporting that misSOD1WT may cause neurodegeneration, expression of hSOD1WT in Tg mice result in a fatal ALS-like disease with neuronal inclusions containing human misSOD1 aggregates.23 Also, in vitro studies in which motor neurons are cocultured with astrocytes derived from patients with ALS suggests an involvement of misSOD1WT in the pathogenesis.24 Collectively, there is emerging evidence that misSOD1WT is neurotoxic and participate in ALS pathogenesis in general and not only in patients carrying mutations in SOD1.
In the present study, we investigated whether inclusions containing misSOD1WT are present in patients carrying mutations in six other ALS-causing genes.
Materials and methods
Human subjects
Since 1993, tissue has been collected at autopsy at Umeå University Hospital, Sweden from patients with ALS and FTD. The EFNS Criteria for Managing ALS25 and the Neary Criteria for FTD26 were used to set the diagnosis. Blood leucocyte DNA from autopsied patients (n=97) was analysed for a panel of ALS-linked genes.12 27 28 Of these patients, 18 had C9orf72HRE (confirmed by Southern blot in CNS tissue),29 2 had FUS mutations, 1 an ALSIN mutation (censored, manuscript in preparation), 1 a VAPB mutation, 1 a NEK1 mutation6 and 11 had SOD1 mutations. Eleven patients carried the coding rs113247976 SNP (P986L) in KIF5A recently found to predispose to ALS, three of these being double mutants C9orf72HRE plus rs113247976 and one A4V SOD1 plus rs113247976 SNP.28
Six additional patients with SOD1 mutations were obtained from Danish and Swiss pathology units. Similar tissues were obtained from 10 control patients with other neurodegenerative conditions and from 20 patients without neurological diseases (table 1, online supplementary table 1).
Supplemental material
Summary of clinical features of the 46 patients with ALS/FTD
All procedures were performed in accordance with the 1964-Helsinki Declaration with amendments and were approved by the Ethical Review Boards for Medical Research at the relevant institutions in Sweden, Denmark and Switzerland. Informed consent for performing autopsies for research purposes was obtained from the patients and/or the next of kin.
Tissue preparation and staining for misSOD1
Tissue samples from the spinal cord (cervical, thoracic and lumbar regions), medulla oblongata, motor cortex, frontal lobe and cerebellum were collected and fixed by immersion in 4% paraformaldehyde in 0.1 M Na phosphate buffer, pH 7.4. After fixation, tissues were embedded in paraffin, sectioned (4 µm), and stained with H&E. The antigen retrieval procedure is essential for detecting misSOD1 and should be mild, since heavily denaturing protocols may cause misfolding of the abundant natively folded SOD1. A well-functional retrieval treatment for sections is 10 min in preheated citrate buffer, pH 6.3, without boiling.18
Immunohistochemical staining (IHC) was initially performed using the ES system and ES reagents and, from 2014, by the more sensitive BenchMark XT automated staining system (both from Ventana Medical System, Illkirch, France) and aminoethylcarbazole or Fast Red was used for development. For immunofluorescent studies, a corresponding fluorescent-labelled secondary antibody was used. Sudan Black diluted to 0.3% was used to avoid autofluorescence. The anti-SOD1 antibodies were diluted to a final concentration of 0.962 µg/mL for 131–153 Ra-ab, 3.7 µg/mL for 57–72 Ra-ab and 1.612 µg/mL for G127X Ra-ab. The sections were examined using a BX53 light microscope (Olympus) and by confocal laser microscopy using a Zeiss LSM 710 confocal microscope and were analysed using the Zen 2011 SP7 software. The presence of intracellular misSOD1 inclusions was rated by two independent examiners according to a 4-point semiquantitative scale.18
Results
Antibodies specifically detect misfolded SOD1
SOD1 is abundant in the CNS constituting 0.1% of the tissue protein.11 17 Carriers of SOD1 mutations show inclusions containing misfolded aggregated SOD1. It is estimated that ≈1% of the total SOD1 protein is contained in such inclusions.11 30 Any misSOD1 inclusions occurring in patients with ALS who lack mutations in SOD1 are likely to be equally scarce. Thus, to detect such species, antibodies that lack reactivity with natively folded SOD1 must be used. Use of Rabbit 1, a Ra-ab raised against native SOD1 with native/denatured reactivity ratio of 0.7/1,31 is found to stain tissues heavily obscuring detection of inclusions containing misSOD1 (online supplementary figure 1C-D). Moreover, the handling of tissue specimen and antigen retrievals have to be lenient to prevent misfolding of native SOD1. We have developed a panel of antibodies, raised against peptides in the human SOD1WT sequence, which only react with disordered or misSOD1 species but show no reactivity against natively folded human SOD1. They were highly specific for SOD1 in western blots of human CNS tissue extracts.17 18 The two primary antibodies used here were rabbit anti-human antibodies (Ra-ab) to misfolded SOD1, raised against peptides corresponding to amino acids (aa) 57–72 and aa131-153. The 57–72 Ra-ab and 131–153 Ra-ab were chosen because they are well characterised17 18 and detect misfolded SOD1 in human tissue studies equally well. For detection of mutated SOD1 in patients with the SOD1 truncating mutation SOD1G127X, we developed a mutant-specific peptide antibody, G127X Ra-ab, directed against the neopeptide sequence unique to this mutation (aa 123-127GQRWK-stop). This antibody reacts exclusively with the mutant protein.11 To test the specificity for misSOD1 in histopathological studies, the 131–153 Ra-ab was preincubated with the immunising peptide in increasing concentrations. This resulted in gradual and finally full disappearance of immunoreactivity (online supplementary figure 1E-H).
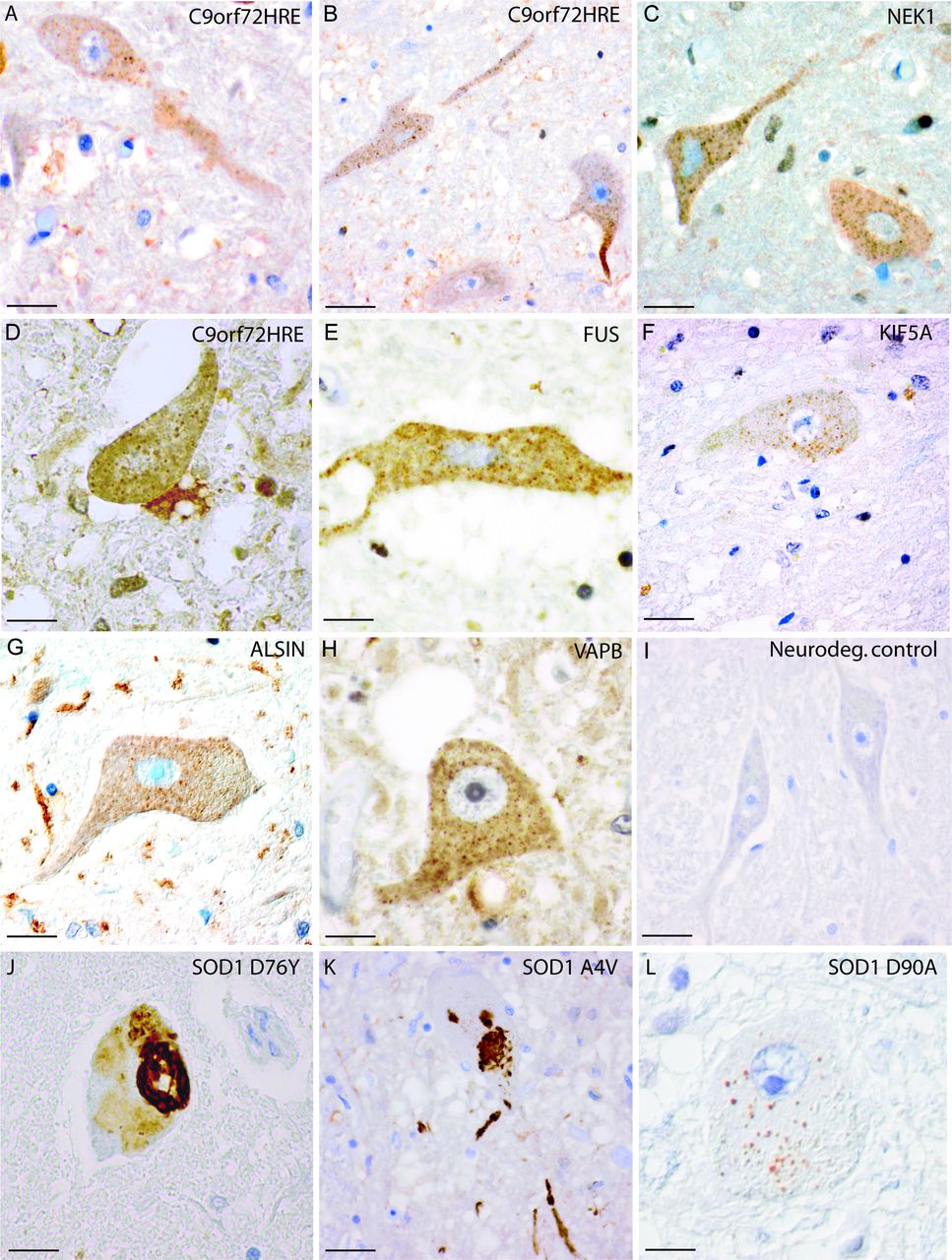
Inclusions of misSOD1WT in spinal cord sections of patients with mutations in six ALS/FTD-associating genes. MisSOD1WT was present in histological sections from patients with ALS carrying C9orf72HRE (A–B, cases #2 and #8), NEK1 mutation (C, case #25), C9orf72HRE mutation (D, case #16), FUS mutation (E, case #27), KIF5A mutation (F, case #19), alsin mutation (G, case #29), VAPB mutation (H, case #26), SOD1D76Y mutation (J, case #37), SOD1 A4V mutation (K, case #31) and SOD1D90A homozygous mutation (L, case #43), but not in a neurodegenerative control case (I, case #47). All sections were stained with the 131–153 Ra-ab peptide antibody. In panels (A–H), multiple small granular cytoplasmic inclusions of misSOD1WT can be seen in motor neurons with a size of approximately 0.5–3 µm. In panel L, staining of the wt-like SOD1D90A mutant is shown. Note the morphological similarity between panel L and panels A–H, where misSOD1WT is stained. In panels J and K, the SOD1 mutations D76Y and A4V, which give rise to unstable SOD1 proteins, are found to show skein-like cytoplasmic SOD1 inclusions in motor neurons. Scale bars: A–F and I 50 µm; G-H, J–L 30 µm. ALS, amyotrophic lateral sclerosis.
Inclusions containing misSOD1WT can be seen in patients carrying C9orf72HRE
Different regions of the brain and spinal cord from 18 patients carrying C9orf72HRE were stained for misSOD1 and analysed. We found small dot-like granular inclusions of misSOD1WT in spinal cord motor neurons in all 18 patients (tables 2 and 3), irrespective of whether the clinical diagnosis was ALS, FTD or both. The inclusions measured 0.5–3 µm were scattered in the cytoplasm of motor neurons and were particularly abundant in the soma (figure 1A,B,D and figure 2F–H) but also present in axons (figure 1A,B). The proportion of spinal motor neurons carrying misSOD1WT inclusions varied between patients; usually, only a few motor neurons per section carried misSOD1WT inclusions. However, in some patients, the proportion was higher. Inclusions were found at all levels of the spinal cord (cervical, thoracic and lumbar) in most patients (table 3). MisSOD1WT inclusions were also present in the neurons of Clark’s nuclei. Furthermore, small inclusions of misSOD1WT were found in the hypoglossus (figure 3D), vagus, facialis and accessorius nuclei. The inferior olivary nucleus did not show misSOD1WT inclusions and was normal in most patients.
Immunohistochemical results of staining for misfolded SOD1 using the misSOD1 peptide antibodies: 131–153 Ra-ab (cases #1–31, #35–56) and G127X Ra-ab (cases #32–34)
MisSOD1 immunohistochemical results
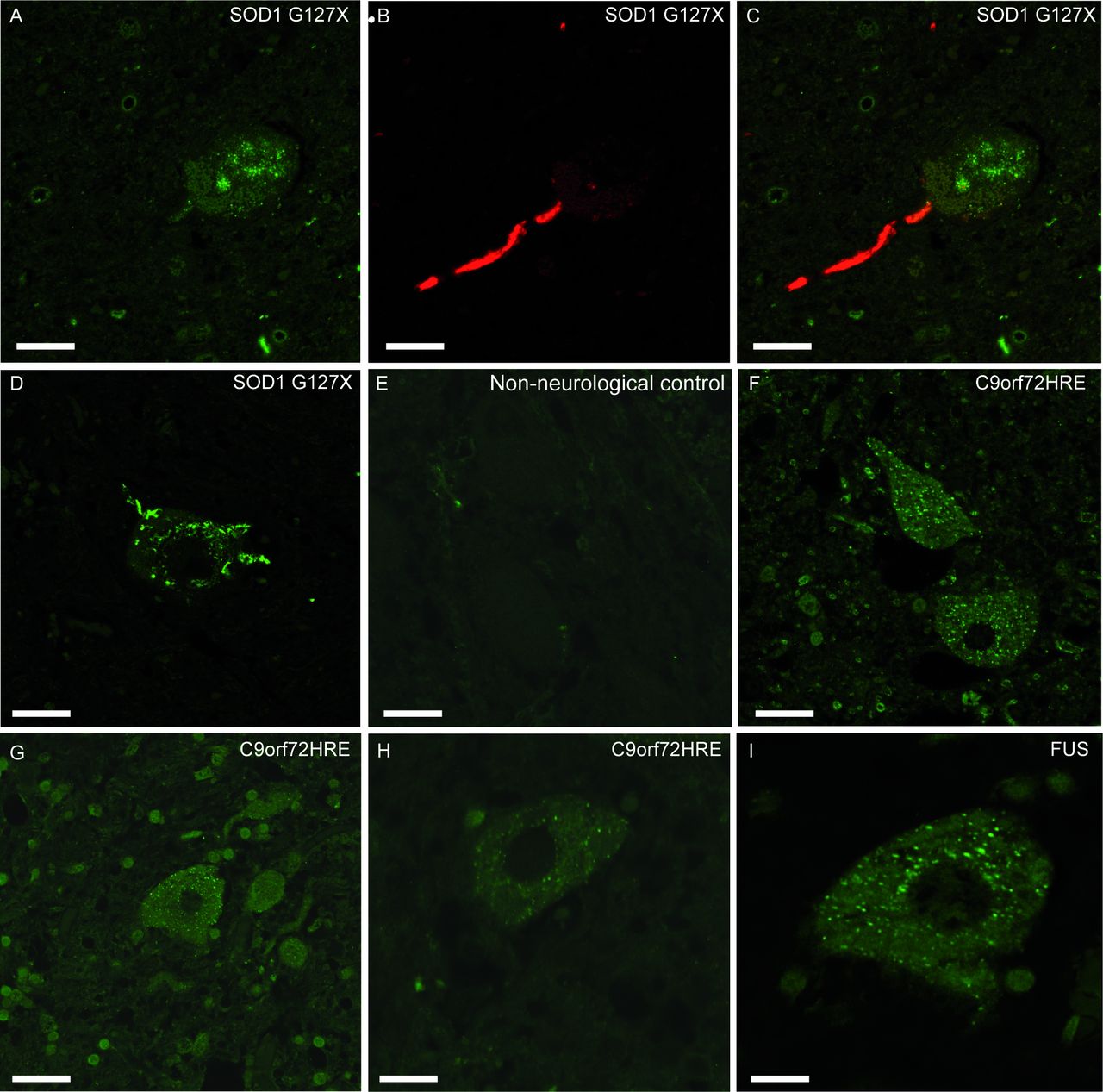
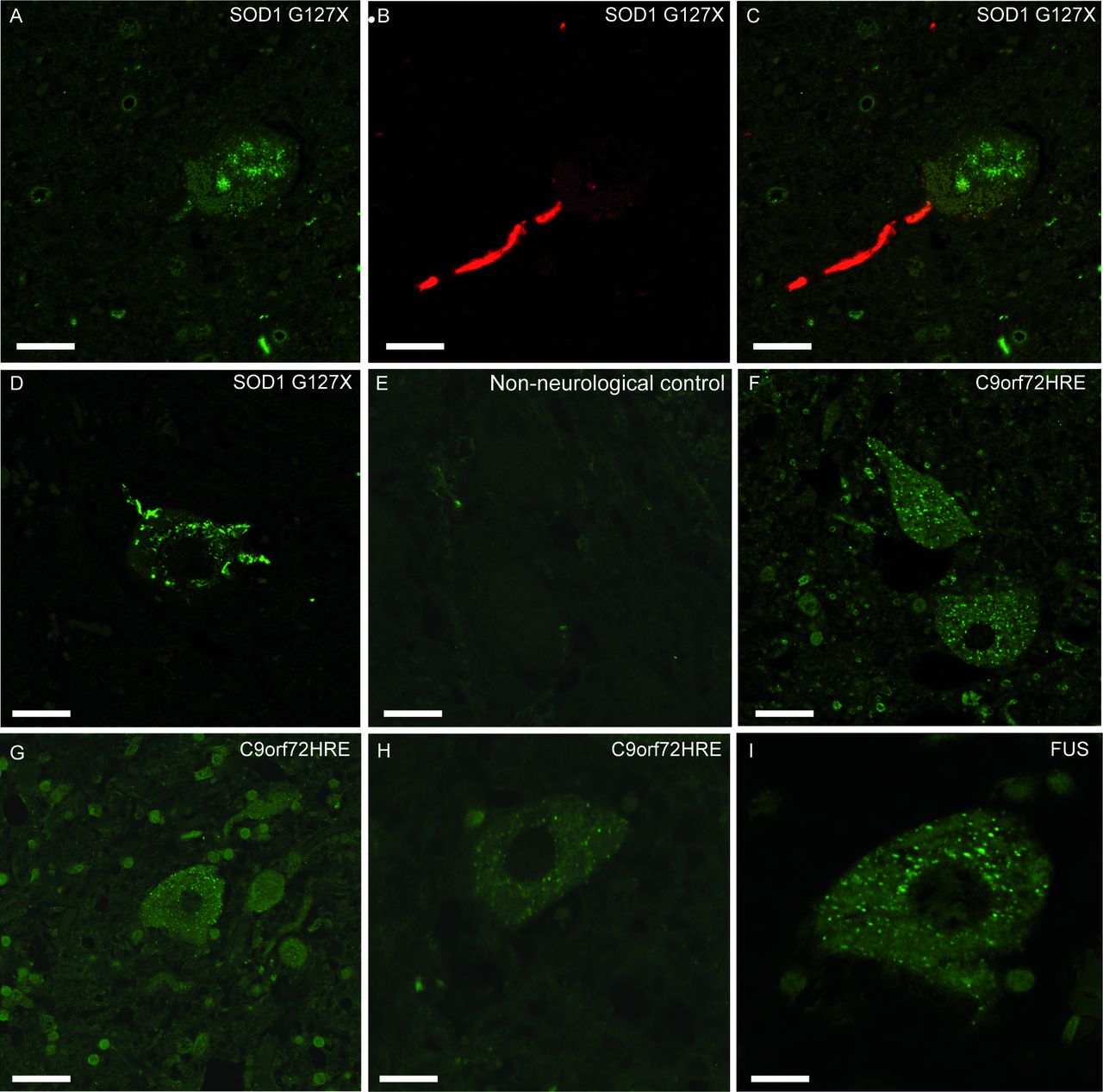
Inclusions of misSOD1 observed in human spinal cord motor neurons. Micrographs showing lumbar ventral horn motor neurons from two patients with the SOD1G127X mutation (A–C, case #33 and D, case #32), a non-neurological control (E, case #64), three patients with C9orf72HRE mutation (F–H, cases #1, #11 and #5) and a FUS mutation (I, case #27). Panels A–C show sections double-labelled with the 131–153 Ra-ab peptide antibody which stains a segment of the SOD1 protein that is absent in the SOD1G127X mutant, and therefore only stains misSOD1WT (A,C, green fluorescence) and the G127X Ra-ab which only recognises the mutated G127X sequence (B,C, red fluorescence). The merged picture of green and red channel scans show that this neuron carries inclusions of both mutated SOD1 (seen here in the axon as red) and of misSOD1WT (seen here in the soma as green). The mutated and misSOD1WT inclusions do not colocalise (C). Panel D was stained with the G127X Ra-ab and panels E–I were stained with the 57–72 Ra-ab peptide antibody. Note the small dot-like granular cytoplasmic inclusions of misSOD1 in motor neurons (F–I) which is not seen in the non-neurological control (E). The mutated, unstable SOD1G127X protein has more of a skein-like appearance (D). Scale bars: A–G, 50 µm, H 30 µm, I 20 µm.

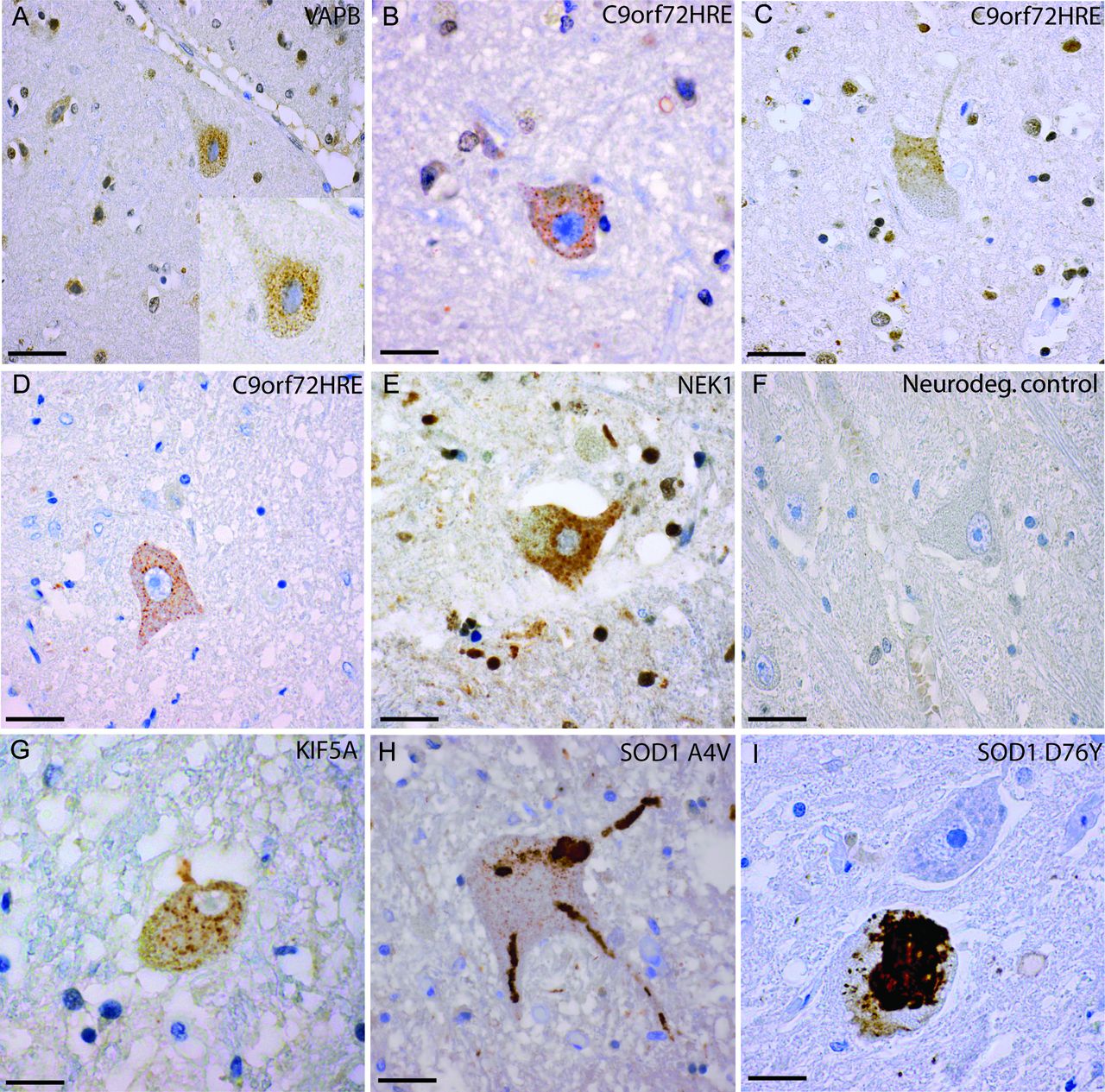
Histological sections from the motor cortex and medulla oblongata of patients carrying different ALS-causing mutations. Sections were stained with the 131–153 Ra-ab peptide antibody. Panels A–C show Betz cells in the motor cortex of patients with a VAPB mutation, (A, case #26) and C9orf72HRE mutation (B, case #13 and C, case #16). Small dot-like cytoplasmic inclusions of misSOD1WT are apparent in the Betz cells (A–C). Panels D–I show neurons in the medulla oblongata of patients with C9orf72HRE (D, case #15), NEK1 mutation (E, case #25), a neurodegenerative control (F, case #55), KIF5A mutation (G, case #19), the SOD1 A4V mutation (H, case #31) and the SOD1 D76Y mutation (I, case #37). In panels D, E and G, inclusions of granular misSOD1 are observed in neurons of the hypoglossal nucleus. Note the mutated SOD1 protein, which appears skein-like in panels H–I compared with the granular misSOD1WT protein (appearing as granular intracytoplasmic dots in panel D (E and G). Scale bars: A 100 µm; B-G 50 µm, H-I 30 µm. ALS, amyotrophic lateral sclerosis.
In the motor cortex, degenerated neurons containing numerous small misSOD1WT inclusions were detected in 7 of 12 C9orf72HRE patients investigated, similar in size to the ones seen in spinal motor neurons (figure 3B,C and table 2) and located both in the cytoplasm and the nucleus. The proportion of motor cortex neurons that carried misSOD1WT inclusions was much smaller than the proportion of spinal motor neurons. In the frontal cortex, 11 out of 16 patients with C9orf72HRE carried misSOD1WT inclusions. Noticeably, in some patients, senile plaques were seen in the motor and frontal cortex, these stained heavily for misSOD1WT.
In the cerebellum of all 12 patients investigated, loss of Purkinje cells and degeneration of white matter was observed. Staining with anti-misSOD1 antibodies revealed misSOD1WT-stained glial cells surrounding either degenerated Purkinje cells or empty baskets. Staining was also found in the neuropil in both grey and white matter as well as in glial cells in the same regions (online supplementary figure 1A, B). In the granular and molecular layers of the cerebellum, p62-positive neuronal cytoplasmic inclusions were observed, a finding proposed to be pathognomonic for this subgroup of ALS.32 No misSOD1WT staining was seen in the granular layer in the patients with C9orf72HRE.
Two of the patients with C9orf72HRE were siblings, one with FTD and the other with ALS (patients 2 and 3, respectively, table 2). The sibling with ALS had a significant loss of spinal motor neurons, whereas the sibling with FTD appeared to have a normal number of spinal motor neurons. However, both had misSOD1WT inclusions of similar morphology in motor neurons at all levels of the spinal cord. No misSOD1WT inclusions were detected in the medulla oblongata. Comparing their frontal cortices, the sibling with FTD had many extracellular senile plaques which stained heavily for misSOD1WT and amyloid-β. This was not seen to the same extent in the ALS sibling. In both, misSOD1WT was rarely observed in cortical glial cell nuclei (table 2).
Patients with FUS, NEK1, KIF5A, VAPB and ALSIN mutations
We found granular inclusions of misSOD1WT in motor neurons of the spinal cord in all patients with FUS, NEK1, KIF5A, VAPB or ALSIN mutations (figures 1C,E–G and 2I and table 2). The misSOD1WT-positive inclusions were morphologically similar to the ones observed in patients with C9orf72HRE and in patients with apparently sporadic ALS.17 Moreover, misSOD1WT pathology in the patients with FUS, NEK1, KIF5A, VAPB or ALSIN mutations included all levels of the spinal cord and brainstem motor nuclei (figure 3E,G), areas of the motor cortex (figure 3A), frontal lobe and the cerebellum. In the patients with FUS mutations, large cytoplasmic FUS-positive inclusions were found in the spinal cord and medulla oblongata when stained with an anti-FUS antibody. In the cerebellum, the same type of histopathology was observed as in the patients with C9orf72HRE, which included staining of misSOD1WT in glial cells of the molecular and granular layers surrounding degenerated Purkinje cells. The patients with VAPB and ALSIN mutations in particular had these changes, whereas the cerebellum in the patients with FUS mutations was less affected. We conclude that all studied patients with FUS, NEK1, KIF5A, VAPB and ALSIN mutations had misSOD1WT inclusions in the spinal cord and brainstem motor nuclei and occasionally also in the motor cortex and frontal lobe.
Comparison with patients carrying different SOD1 mutations
For comparison, we stained tissue from 17 patients carrying SOD1 mutations (tables 2 and 3). The six patients homozygous for the SOD1D90A mutation had small granular inclusions (figure 1l) morphologically similar to the inclusions described above in patients carrying mutations in C9orf72HRE, FUS, NEK1, KIF5A, ALSIN and VAPB and previously in sALS.17–19 22 This could be expected since the SOD1D90A mutation produces an essentially fully stable SOD1D90A protein with biophysical properties of SOD1WT.33 All patients with the SOD1D90A mutations had mild cortical atrophy with neuronal loss, reactive gliosis and microvacuolisation of the three superficial cortical laminae. MisSOD1D90A inclusions in the motor cortex and frontal lobe were rarely observed in these patients. In contrast, staining for misSOD1 in patients with ALS heterozygous for the mutations A4V, V5M, D76Y, D83G, D101G, G114A, L144F and G127X - all encoding unstable SOD1 mutants11 12 34 revealed larger skein-like and Lewy body-like inclusions in addition to smaller granular misSOD1 inclusions (figures 1J,K–3H,I). Thus, there are (at least) two different morphologies of misSOD1 inclusions present in patients carrying pathogenic mutations in SOD1.
The SOD1G127X mutation results in a shift in the DNA coding frame and a neopeptide sequence of five amino acids 127GQRWK-stop, truncating after codon 132, 21 amino acids short of end of the SOD1WT polypeptide.11 It is therefore suited for studying different misSOD1 inclusions. Staining with the anti-SOD1 antibody raised against the C-terminal end (131–153 Ra-ab) revealed small and dot-like misSOD1 positive inclusions in the motor neurons that resembled the staining seen in patients with sporadic and familial ALS lacking SOD1 mutations (figure 2A). This was observed in all three patients heterozygous for this mutation. Double staining with the 131–153 Ra-ab and the mutation-specific antibody G127X Ra-ab revealed separate inclusions of misSOD1 in motor neurons, without colocalisation (figure 2A–C): the large skein-like inclusions contain the misfolded SOD1G127X mutant protein, the small granular dot inclusions the misSOD1WT generated from the wild-type allele. Both types of inclusions in the same motor neuron were only observed in a few motor neurons per section. Most motor neurons had only one type of SOD1 inclusion and a few neurons lacked inclusions.
Control patients and reanalysis of spinal cord sections from patients with ALS and controls
To determine whether the misSOD1WT inclusions were specific for patients with ALS and FTD, brain and spinal cord tissue from 10 control patients with other neurodegenerative diseases were stained with the same anti-misSOD1 antibodies. Nine of 10 controls did not show misSOD1WT inclusions anywhere in the spinal cord or brain (table 2, figures 1I and 3F). A single patient diagnosed with epilepsy after a cerebral infarction had some misSOD1WT inclusions in the spinal cord and hypoglossal nucleus. Neuronal cytoplasmic misSOD1WT inclusions were also seen to the same extent as in the motor cortex of patients with ALS, but the frontal cortex and cerebellum were blank. We additionally analysed spinal cord sections from cervical, thoracic and lumbar regions of 20 patients with non-neurological diseases (online supplementary table 1) with the anti-misSOD1 antibodies. No inclusions of misSOD1WT were observed in any of these (figure 2E).
The above reported IHC investigations were carried out over a period of 14 years. During this time, the staining equipment was changed in our lab. For control purposes, we recently restained and re-examined in parallel lumbar spinal cord specimen from all 30 controls and 38 (of the 46) patients with ALS at the same time (CNS tissues were no longer available from two patients with C9orf72 and six patients with SOD1 mutations). Two different antibodies, 57–72 Ra-ab and 131–153 Ra-ab—targeting the active site and the carboxy end of the SOD1 protein, respectively—were used to ascertain the specificity for misSOD1 in the stainings. Identical protocol was used for all specimen. The identities of the restained sections were coded and were independently examined by two neuropathologists (KF, TB).
The results are summarised in figure 4, online supplementary figure 2 and supplementary table 2. The two examiners assessed the sections in principle equally, the only differences being a few one step differences in gradings of the SOD1 stainings in patients with ALS, online supplementary table 2. In these cases, both gradings are shown. All the patients with ALS without SOD1 mutations again showed the misSOD1WT-positive granular inclusions. They were easily detected and had the same shape and locations as seen in the previous older stainings. The misSOD1WT staining in the neurodegenerative control #50 was again seen (online supplementary figure 2 AI). However, the principal difference from the previous analyses was that misSOD1WT staining was now seen in three more neurodegenerative control (online supplementary figure 2 AG, AH, AK), which did not appear in the previous stainings (table 2). Two of these patients had Parkinson’s disease and one had Alzheimer’s disease. Noticeably, only a few motor neurons were positive. Moreover, also two of the non-neurological controls were when restained now graded as positive since a few motor neurons were positive (online supplementary figure 2 AQ, AW).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Restaining of misSOD1WT inclusions in lumbar spinal cord sections from patients carrying different ALS/FTD associated gene mutations as well as neurodegenerative and non-neurological controls. Micrographs show (A,B) C9orf72HRE (case #15), (B,C) FUS (case #27), (E,F) non-neurological control (case #71), (G,H) non-neurological control (case #72), (I,J) KIF5A (case #22), (K,L) alsin (case #29), (M,N) VAPB (case #26), (O,P) NEK1 (case #25), (Q,R) neurodegenerative control (case #54), (S,T) neurodegenerative control (case #55), U-V SOD1A4V (case #31), W-X SOD1D90A (Case #43). For comparison, sections were stained with either the 131–153 Ra-ab (A, C, E, G, I, K, M, O, Q, S, U and W) or the 57–72 Ra-ab (B, D, F, H, J, L, N, P, R, T, V and X). Multiple small dot-like inclusions are seen with both antibodies in all patients carrying different ALS/FTD associated gene mutations but are absent in the neurodegenerative and non-neurological controls. Note the high background staining when using 57–72 Ra-ab. Scale bars: A-X 100 µm for all micrographs.
The outcomes of examinations of sections stained with two different anti-SOD1 peptide antibodies (57–72 Ra-ab, 131–153 Ra-ab) were almost identical (figure 4 and online supplementary table 2). This proves that the component that becomes stained in the granules is indeed misSOD1WT. The 131–153 Ra-Ab gives generally less background staining and was therefore chosen for most of the histochemical panels shown.
Discussion
We here report the finding of misSOD1WT-positive intracytoplasmic inclusions in motor neurons of patients lacking SOD1 mutations but carrying pathogenic mutations in six other ALS-causing genes C9orf72HRE, FUS, NEK1, KIF5A, ALSIN and VAPB. The inclusions were small, numerous and scattered in the cytoplasm of motor neurons at all levels of the spinal cord, in the motor nuclei of the brain stem and occasionally in the motor and frontal cortex. They were seen in all 18 patients carrying C9orf72HRE, regardless of whether the patient had a ‘pure’ FTD diagnosis, pure ALS diagnosis or both. The morphology and shape of these inclusions resembled that of the inclusions described in sALS.17 Patients with other FTD-associated and ALS-associated mutations had similar inclusions that stained for misSOD1WT in the brain and spinal cord. The finding that misSOD1WT appears in small inclusions in all studied patients with ALS raises the question if these misSOD1WT inclusions are different from the ones formed from mutated SOD1.
In SOD1 Tg murine models overexpressing the G85R, G93A, D90A or G127X human SOD1 mutations, larger skein-like inclusions are observed when stained with human misSOD1-specific antibodies. The misSOD1 in these SOD1 Tg murine models is aggregated. So far, two strains of aggregates (A and B) have been described which differ with regard to molecular structure.14 Both display prion-like properties and propagate further aggregation and phenotypically different ALS-like diseases when inoculated into the spinal cord of 100-day-old asymptomatic mice Tg for the human SOD1G85R mutation.15 In patients that carry unstable mutant SOD1s, morphologically similar misSOD1-IR inclusions are found.11 These also appear to contain aggregated SOD1, but the molecular structures have yet to be elucidated. Inoculation of SOD1G127X aggregates purified from the spinal cord of patient no. 32 in this study (table 1) into the lumbar spinal cord of 100-day-old mice induced hSOD1-aggregate formation throughout the neuraxis concomitant with onset of ALS.16 Binary epitope mapping revealed that the human misSOD1 aggregates was of the A-type.16 Further inoculation studies of SOD1 aggregates purified from other patients from this study are ongoing.
In patients homozygous for the stable wt-like mutant SOD1D90A, the motor neurons display small granular misSOD1D90A inclusions. Whether these also have prion-like features as the larger inclusions found in the murine Tg models and in patients that carry unstable mutant SOD1s, remains to be determined. Still, this is the sole conspicuous misSOD1-IR structure found in postmortem tissue from the six SOD1D90A homozygous patients. The differences in morphology compared with patients with other SOD1 mutations might be related to their different molecular stabilities. SOD1WT and stable SOD1 mutants might associate to form small granular inclusions whereas unstable mutant proteins could either be rapidly degraded or form fewer but larger inclusions. Interesting implications follow from this observation: (1) inclusions of misSOD1 may be pathological hallmarks in all forms of ALS and (2) variation in morphology, distribution and number of aggregates may be used to distinguish different forms and stages of ALS.
Summarising the results of the initial staining and the restaining, 4 of 10 neurodegenerative controls had some inclusions of misSOD1WT (online supplementary figure 2 AG-AI, AK, and supplementary table 2). One of the positive controls was a patient with epilepsy after cerebral infarction. Here, small granular cytoplasmic inclusions of misSOD1WT were seen in spinal cord motor neurons (online supplementary figure 2 AI), in the hypoglossus nuclei and in the motor cortex. No inclusions were found in the striatum, frontal cortex or mesencephalon. Speculatively, this control individual could have had clinically unrecognised early ALS and/or the cerebral infarction triggered a cascade of events resulting in misfolding of misSOD1WT that might not have prion-competent conformations15 but are still detected by our polyclonal antibodies. However, cerebrovascular injury has been proposed to be a risk factor for ALS.35 Two of the positive controls had Parkinson’s disease (online supplementary figure 2 AG, AK). Interestingly, inclusions of misSOD1WT have been found to accumulate in the brains of patients with Parkinson’s disease and suggests common mechanisms of misSOD1WT aggregation in both disorders.36 Moreover, neurodegenerative diseases with concomitant copathology is frequent.37
Two of the non-neurological controls also had a few motor neurons that stained positive for SOD1. One died of a dissection of the aorta and the other of myocardial infarction (online supplementary figure 2 AQ, AW). The first showed small areas of infarction in the spinal cord. As also noted earlier in senile plaques, the state of the tissue might thus affect the immunoreaction found. All other 24 controls stained negative for misSOD1 in all studied tissue specimens, demonstrating major differences between patients with ALS and controls.
The presence of misSOD1WT in postmortem CNS tissue of sALS and fALS without SOD1 mutations is controversial: some studies support the result of the present study,17–22 but in other studies misSOD1 could not be detected in sALS38–40 or there were no differences between sALS and control individuals.41 42 Reasons for these opposite results were discussed in a recent article22 and includes methodological differences (ie, the use of TRIS/EDTA buffers instead of citrate-based buffers, different preanalytical tissue handling, variable incubation times, different antibody dilutions). Guidelines for staining misSOD1 were proposed.22 These guidelines were adhered to in this work. It should be noted that Ref. 21 was a blinded study of sALS cases compared with controls (some of these were the same as in this study) and using some of the anti-misSOD1 antibodies as in the present study.
The present morphological study does not prove that misSOD1WT causes neurodegeneration in patients carrying mutations in C9orf72HRE, FUS, NEK1, KIF5A, ALSIN or VAPB. Nor can we explain how patients with a massive intronic GGGGCC repeat-expansion in C9orf72 or a R495X-truncation in the mRNA-housekeeping FUS enzyme have inclusions of misSOD1WT in their motor neurons and with a morphology and similar pattern of cellular distribution as in patients with the SOD1D90A mutation. We speculate that whatever the initial pathogenic events are in patients carrying C9orf72HRE or coding mutations in FUS, NEK1, KIF5A, VAPB or ALSIN, eventually they converge to common downstream event(s) involving unfolding and misfolding of SOD1WT. For this to happen, the stabilising intrasubunit C57-C146 disulfide bond must be reduced and the stabilising Zn2+ cofactor released. If the generated misfolded SOD1 species are pathogenic and form SOD1WT prions, based on injection of SOD1 aggregates,15 16 this event may only have to occur in a limited number of cells, perhaps only once for the SOD1-prion cascade to be initiated. The finding that overexpression of human SOD1WT in mice elicits a fatal ALS-like disease supports that SOD1WT can cause neurodegeneration.23 The observation that all three patients heterozygous for the SOD1G127X mutation had inclusions with the truncated mutant SOD1G127X protein (easily recognisable because of its neopeptide sequence) and separate inclusions containing only misSOD1WT (detectable by the amino acid sequence that is missing in the mutated SOD1 protein) is interesting. It raises the possibilities that either the misfolding of SOD1WT is a secondary non-pathogenic event in a stressed cell or that misfolded SOD1WT participates and could be pathogenic per se, raising the possibility that there could be more than one kind of SOD1 prions active simultaneously in patients with SOD1 mutations. Simply targeting a mutant SOD1 gene with tailored genetic inhibition therapy may therefore not be sufficient to halt disease progression. This observation is important, since new therapies targeting SOD1 are ongoing or are being planned. If SOD1 plays a role in subtypes of ALS without SOD1 mutations as our results suggest, then many patients may benefit from these new anti-SOD1 therapeutics.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Acknowledgments
We are indebted to all the patients who made this study possible. The authors wish to thank Ulla-Stina Spetz, Eva Bern, Helena Alstermark, Ann-Charloth Nilsson, Eva Jonsson, Agneta Öberg and Matthew Marklund for technical assistance.
References
Footnotes
TB and PMA are joint senior authors.
Contributors Study concept: PA. Designed the study: KF, SM, TB, PA. Collected and evaluated the material: KF, KG, BP, MW, MN, TB, PA. Performed and evaluated the laboratory analysis: KF, MW, MN, SLM, TB. Wrote the manuscript: KF, TB, PA.
Funding This work was supported by grants from the Swedish Brain Foundation (grants nos. 2012-0262, 2012-0305, 2013-0279, 2016-0303), the Swedish Science Council (grants nos. 2012-3167, 2017-03100), the Knut and Alice Wallenberg Foundation (grants nos. 2012.0091, 2014.0305), the Bertil Hållsten Foundation, the Ulla-Carin Lindquist Foundation, the Kempe foundation, the Neuroförbundet Association, the Torsten and Ragnar Söderberg Foundation, Umeå University Insamlingsstiftelsen (223-2808-12, 223-1881-13, 2.1.12-1605-14) and the Västerbotten County Council (grants nr. 56103-7002829).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the Regional Medical Ethical Review Board in Umea, Sweden with applications from 1994 (FEK no. 94-135) and 2014 (EPN no. 14-17-31M).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement There are additional pictures of misSOD1+ stainings available from the here studied material which we are willing to share.