Article Text

Abstract
Objectives Chronic inflammatory axonal polyneuropathy (CIAP) is defined on the basis of the clinical, electrophysiological and nerve biopsy findings and therapeutic responses of ‘immunotherapy responding chronic axonal polyneuropathy (IR-CAP)’.
Methods The diagnosis of IR-CAP was made when all of three of the following mandatory criterion were met: (1) acquired, chronic progressive or relapsing symmetrical or asymmetrical polyneuropathy with duration of progression >2 months; (2) electrophysiological evidence of axonal neuropathy in at least two nerves without any evidence of ‘strict criteria of demyelination’; and (3) definite responsiveness to immunotherapy.
Results Thirty-three patients with IR-CAP showed similar clinical features of chronic inflammatory demyelinating polyneuropathy (CIDP) except ‘motor neuropathy subtype’. High spinal fluid protein was found in 27/32 (78%) cases. ‘Inflammatory axonal neuropathy’ was proven in 14 (45%) of 31 sural nerve biopsies.
Discussions IR-CAP could well be ‘axonal CIDP’ in view of clinical similarity, but not proven as yet. Thus, IR-CAP is best described as CIAP, a distinct entity that deserves its recognition in view of responsiveness to immunotherapy.
Conclusion Diagnosis of CIAP can be made by additional documentation of ‘inflammation’ by high spinal fluid protein or nerve biopsy in addition to the first two diagnostic criteria of IR-CAP.
Statistics from Altmetric.com
Introduction
An axonal Guillain-Barré syndrome (GBS) has been well established as a separate entity.1 An ‘axonal chronic inflammatory demyelinating polyneuropathy’ (CIDP) has been rarely been reported in 1990s.2 However, there has been some doubt as to the existence of such an entity, ‘axonal CIDP’ or chronic inflammatory axonal polyneuropathy (CIAP). In this article, we report our experience on 33 cases of ‘immunotherapy responding chronic axonal polyneuropathy (IR-CAP)’ at the University of Alabama at Birmingham (UAB) and present the nerve biopsy evidence of inflammatory axonal neuropathy in this entity. We think that this entity represents CIAP and finally present proposed diagnostic criteria of CIAP.
Materials and methods
The diagnosis of IR-CAP was made when all of three of the following mandatory criteria were met: (1) acquired, chronic progressive or relapsing symmetrical or asymmetrical polyneuropathy with duration of progression >2 months; (2) electrophysiological evidence of axonal neuropathy in at least two nerves without any evidence of ‘strict criteria of demyelination’; and (3) definite responsiveness to immunotherapy.
Electrophysiological criterion of axonal neuropathy is defined as abnormal nerve conduction study (NCS) without any electrophysiological criterion of demyelination in any motor or sensory NCSs in box 1.3–5
Electrophysiological criteria of demyelination*
Motor nerve conduction
Nerve conduction velocity (NCV): <40% of normal mean
Terminal (distal motor) latency: >150% of normal mean
F-wave latency: >150% of normal mean
Conduction block: >50% decrease in compound muscle action potential (CMAP) amplitude (total or negative peak)
Dispersion: distal and proximal dispersion by the negative peak or total duration of the CMAP:
Distal: >8 ms for negative peak duration or 35 ms for total duration
Proximal: >30% for negative peak duration or >33% for total duration compared with distal CMAP.
Sensory nerve conduction
Negative peak NCV: <40% of normal mean
Dispersion: duration of compound nerve action potential: >2.8 ms with surface electrode recording or >4.8 ms with near-nerve needle recording
To assess the degree of disability, we used the disability scoring system for peripheral neuropathy. According to this system, 0 is normal and 20 is maximally disabled. The disability score is the total sum of four different modalities: two motor functions (distal and proximal) and two sensory functions (pin-prick and touch, vibration and position).6 Improvement is defined when there is an objective improvement by at least 2 ‘disability scores’ within 1 year of treatment.
Routine nerve studies (NCS) were performed using standard surface e electrode placement as previously described. All patients underwent motor NCS in median, ulnar, fibular and tibial nerves, sensory NCS in median, ulnar and sural nerves, and mixed NCS in median and ulnar nerves. Monopolar needle EMG was performed in all patients in muscles chosen by EMGer. In one patient, the on-nerve needle sensory NCS of the sural nerve was performed.5
In 32 patients, the CSF was obtained by lumbar puncture for diagnostic purposes. All patients had basic peripheral neuropathy workups that included thyroid and rheumatology profiles, vitamin B12 and folic acid, haemoglobin A1C, sedimentation rate and immunoelectrophoresis of serum protein by immunofixation.
Sural nerve biopsy was performed using the standard method behind the lateral malleolus and studied in the frozen, paraffin and semithin sections in 31 patients.7 Axonal degeneration is determined by myelin-digestion chambers on the frozen sections. Inflammatory cells are assessed on the frozen and paraffin sections. Demyelination is assessed on the semithin section.
The clinical and laboratory features of IR-CAP were compared with those features of CIDP previously published.8
Results
Diagnosis
During 25 year period from 1990 to 2014, we identified 39 patients with chronic axonal polyneuropathy who were treated with immunotherapy. Among 39 patients, we identified 33 patients with IR-CAP, in which all criteria 1, 2 and 3 are met. Six patients were excluded because of lack of any response to immunotheraphy over 3–9 years period. Among these six patients, one had progression over 4-year period even with continued treatment with monthly IVIG together with various combination of cychlorphosphamide, mycophenolate monfetil and ciclosporin. In other five cases, their neurological status was stabilised with mainly with IVIG and azathioprine over 0.5–6 years period.
Clinical features of IR-CAP
Five cases had previous episode of neuropathy prior to our evaluation (table 1). One had diagnosis of axonal GBS 4 years before the first relapse of neuropathy. One had two episodes of double vision and neuropathy over 7 year’s period.9 One had one episode of neuropathy which was dramatically improved with steroid by a referring neurologist. Two cases had previous episode of spontaneously improved neuropathy. Antecedent event or infection in 4–8 weeks prior to the onset was observed in eight cases: flu-like episode in four, diarrhoea in one, flu-shot in one, hernia surgery in one and herpes zoster in one. Symmetrical motor-sensory neuropathy was the most common form of neuropathy in 26 cases. Only one case had symmetrical sensory neuropathy. In 22 cases, proximal and distal muscles of limbs were involved in neuropathy while distal muscles solely in eight cases and proximal muscles in three cases. Sensory abnormality was found in distal limb area in 25 cases and in proximal limb area in 7 cases. Astreognosis was present in two cases. Ataxic gait was observed in three cases. Atrophy was observed in hands in two and in legs in three cases. In six cases, cranial nerves were impaired: total ophthamoplegia in onepartial ophthalmoplegia with facial numbness in one, aniscoria in one, facial diplegia in one, swallowing difficulty in one and breathing difficulty in two. Except one case with an increased knee reflexes, all cases had reduced or absent reflexes in one of four tested reflexes. Fasciculation was not observed in any case.
Comparison of clinical and laboratory features between immunotherapy responding chronic axonal polyneuropathy (IR-CAP) and chronic inflammatory demyelinating polyneuropathy (CIDP)
Laboratory findings
All routine workups for peripheral neuropathy were negative except paraproteinemia in five cases, and low immunoglobulin in 1. Serum autoantibody neuropathy panel including GM1 was checked in 12 cases. One case with symmetrical motor-sensory neuropathy had positive MAG antibody without any monoclonal gammopathy at onset and negative MAG antibody in 3 years. One case mimicking chronic ataxic neuropathy ophthalmoplegia M-protein agglutination disialosyl antibodies (CANOMAD) syndrome had monosialosyl antibodies.8 9 Thus, associated diseases were found in six cases: monoclonal gammopathy in three, biclonal gammopathy in one, hypogammoglobulinopathy in one and multiple myeloma in one.
The most prominent laboratory abnormality was elevated CSF protein (>55 mg/dL) in 25 (78%) of 32 tested cases. None of cases showed any increase in cell. IgG level was increased in four cases.
Electrophysiological findings
NCS was abnormal in all cases. All cases had axonal neuropathy. None of cases met the criteria of demyelination by the UAB criteria or Joint Task Force of the European Federation of Neurological Societies (EFNS)/Peripheral Nerve Society (PNS) Guideline.10 The most prominent neuropathy pattern in NCS was mixed motor and sensory neuropathy in 28 cases. Three cases had motor neuropathy pattern (pure motor NCS abnormality). Sensory neuronopathy pattern indicative of sensory ganglionopathy was observed in one case. One case had sensory neuropathy pattern.
Motor NCS was abnormal in 31 cases. The most common motor nerve conduction abnormality was absent or low CMAP amplitude in 51 (39%) of 132 tests. Slow NCV was observed in 48 (36%) tests while prolonged F-wave and terminal latencies, in 49 (37%) and 42 (32%) tests each. F-wave was absent in 17/121 (14%) tests. Percentage increases above mean values of F-waves were below 22% in all four nerves.
Sensory NCSs of median, ulnar and sural nerves were abnormal in 28 cases. The most common abnormality was slow NCV in 62 (63% of 99 tests followed by low sensory compound nerve action potential (CNAP) amplitude 48 (48%) of 99 tests and absent potential in 40 (40%) of 99 tests. Absent sensory CNAP was most common in sural nerve (23 cases) in contrast to median (5 cases) and ulnar (5 cases) nerves. ‘Sural sparing’ (abnormal median or ulnar and normal sural sensory nerve conduction) was observed in two cases.
Mixed NCSs of forearm and upper arm segments of median and ulnar nerves were abnormal in 25 cases. The most common abnormality was low amplitude of mixed CNAP in 15 (23%) of 66 tests followed by slow mixed NCV in 9 (14%) of 66 tests and absent mixed CNAP in 4 (6%) of 66 tests. Near-nerve needle (NNN) sensory nerve conduction in the plantar nerve showed absent potential in two cases and normal finding in one case. On-nerve needle nerve conduction in the sural nerve in one case showed ‘axonal neuropathy’.5
Fibrillation and positive sharp waves were detected in distal muscles in 26 (89%) of 29 tested cases, in proximal muscles in 12 (86%) of 14 tested cases, and in lumbosacral paraspinal muscles in 11 (85%) of 13 tested cases.
MRI scan of the roots and plexus
One patient had ‘christmas light pattern’ in the lumbosacral area and a high CSF protein. She had complete recovery with a long-term steroid treatment.
Nerve biopsy findings
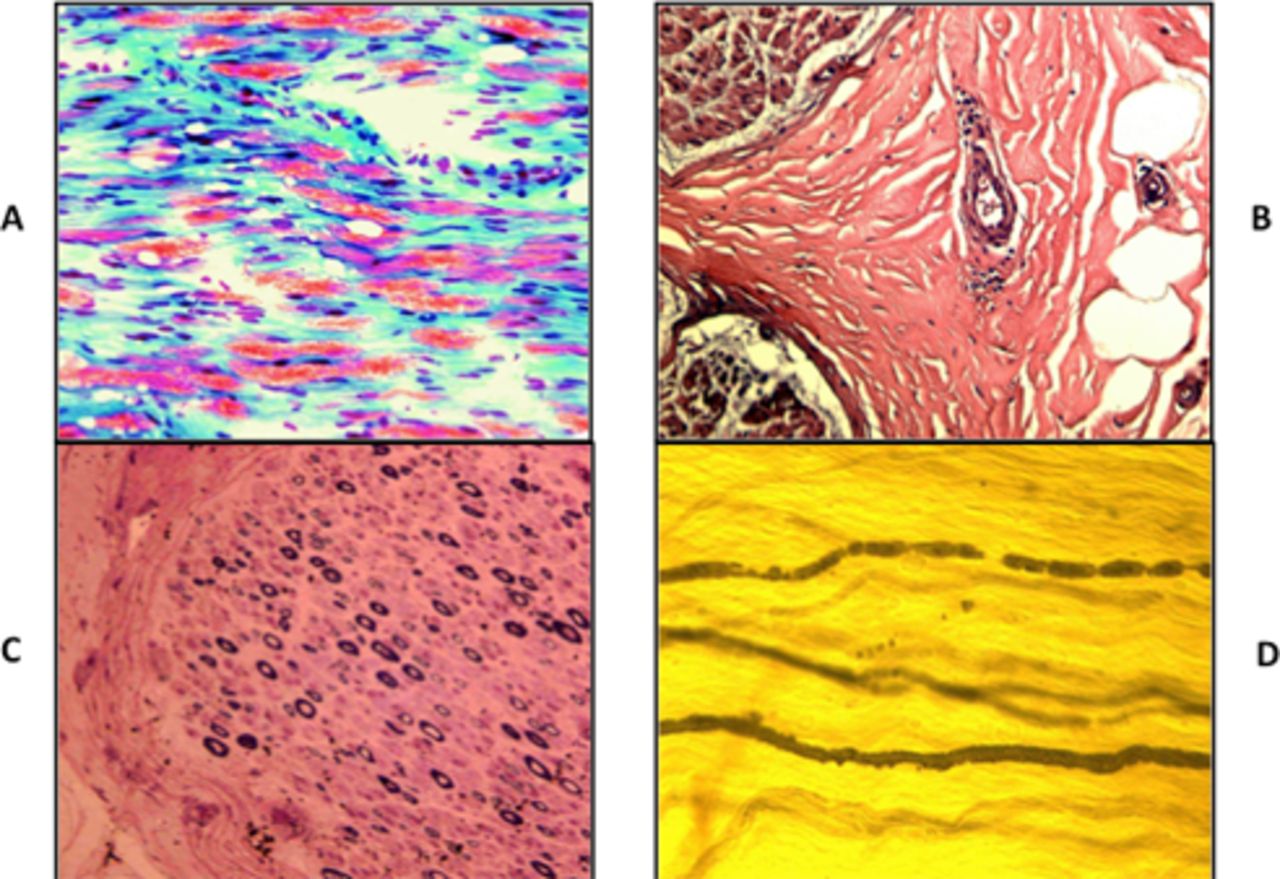
Sural nerve biopsy in 31 cases showed normal finding in one case. The most prominent finding was inflammatory neuropathy in 14 (45%) cases with axonal degeneration in 11 (36%) cases. Inflammatory cells were seen in the epineurial perivascular areas (figure 1). The next most prominent finding was axonal neuropathy demonstrated by myelin-digestion chambers in the modified trichrome stain in the frozen sections in 16 (51%) cases. Teasing of nerve in two cases showed many myelin ovoids typical of axonal degeneration.
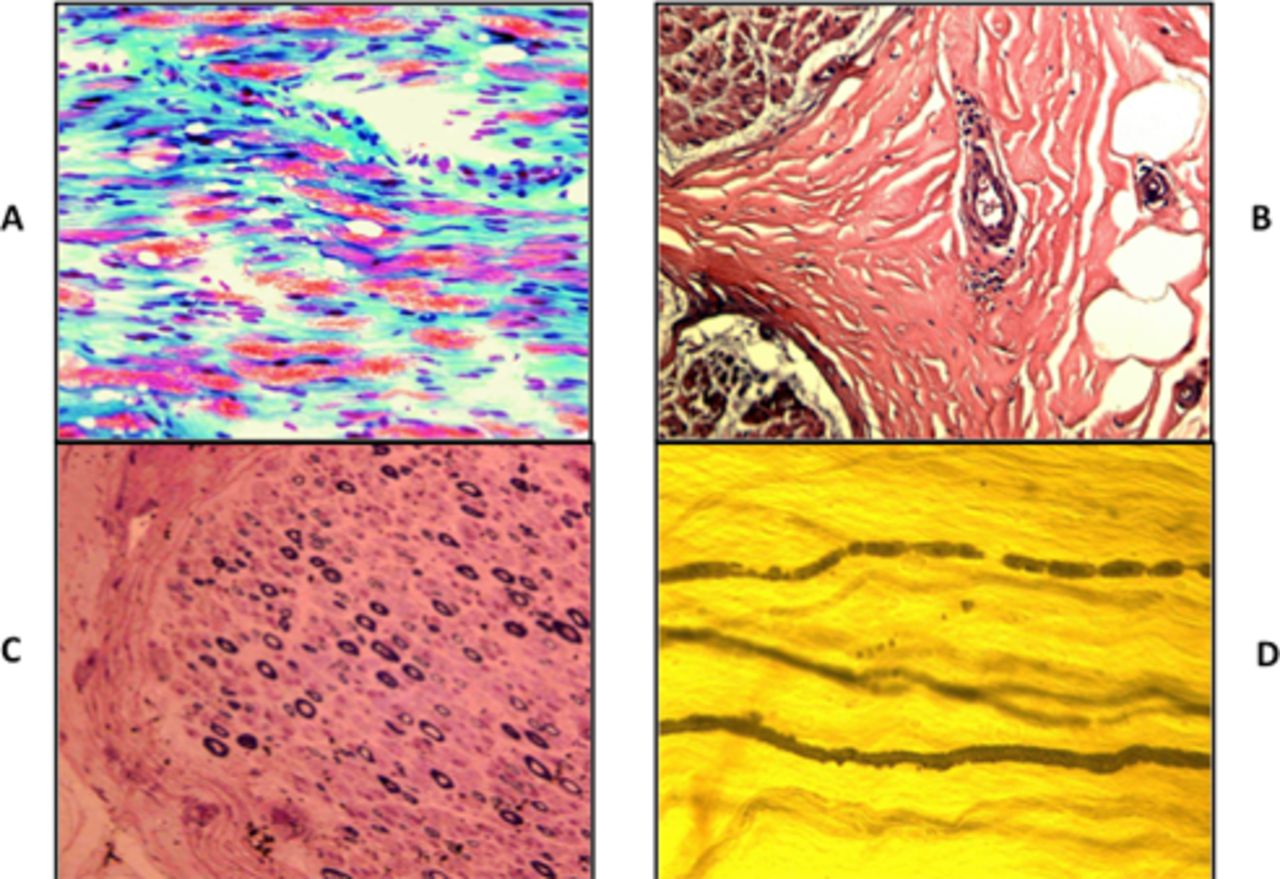
Sural nerve biopsy findings in chronic inflammatory axonal polyneuropathy. (A) Modified trichrome stain. Frozen section. ×200 Prominent myelin-digestion chamber representing active axonal degeneration. (B) H&E stain ×200 perivascular collection of inflammatory cells in the epineurial space. (C) Semithin section. Toluidine blue and basic fuchsin stain (Paragon multiple stain) × 200 moderate axonal loss with no evidence of onion bulb formation or thinly myelinated fibre. (D) Nerve fibres teasing. Osmic acid stain. × 200 Myelin ovoids representing axonal degeneration.
Treatment response and outcome
Spontaneous improvement was not observed in any of cases. In all cases, the various immunotherapy was tried. Clinical improvement was documented in 33 cases by design. Maximal clinical improvement was achieved with treatment over 1 week to 1 year period, within 6 months in 90% of cases. Prednisone therapy was the main treatment in 20 cases. Azathioprine was the next commonly used immunosuppressor medication in 15 cases. IVIG was the most commonly added treatment in 16 cases. Plasma exchange was only used in two cases. Ciclosporine, mycophenolate mofetil and methotrexate were used as adjunctive medication in three cases. Follow-up treatment response during 6 months to 20 years was observed in 29 cases. In 29 cases, the improved status was maintained with prednisone in 14 cases, azathioprine in 14, IVIG in 12 various combination treatments. Relapse was documented in 14 (42%) cases.
Comparison between IR-CAP and CIDP
The only significant difference (p=0.0057) between IR-CAP and CIDP was ‘pure motor neuropathy’, being more common in CIDP. Otherwise, IR-CAP had many similarities of clinical, laboratory and therapeutic characteristics of CIDP (table 1).
Terminal (distal motor) latencies and motor NCVs were compared between IR-CAP (CIAP) (figure 2). Mean terminal latencies in four motor nerves are 21%–33% above normal mean in IR-CAP and 109%–135% above normal mean in CIDP, beyond ‘demyelination marker’ (>50% of the normal mean). Mean NCVs in four nerves are 0%–23% decrease of normal mean in IR-CAP and 33%–38% decrease of normal means in CIDP, near to demyelination markers (>40% below the normal mean) in CIDP. Thus, comparison of terminal latencies and NCSs clearly showed that abnormal nerve conduction in IR-CAP is ‘axonal’ compared with ‘demyelination’ in CIDP.

{kind=link}
{kind=link}
Comparison of terminal (distal motor) latency and nerve conduction velocity between chronic inflammatory axonal polyneuropathy (CIAP) and chronic inflammatory demyelinating polyneuropathy (CIDP). Terminal latency: mean terminal latencies in the median, ulnar, fibular and tibial nerves. 0% represents normal (N) means. >50% (red line, 50% beyond normal means, represents demyelination). Nerved conduction velocity: mean nerve conduction velocities in the median, ulnar, fibular and tibial nerves. 100% represents normal (N) means. <60% (red line; 40% below normal means) represents demyelination. D distal (elbow–wrist) segment. P, proximal (axilla–elbow) segment.
By definition, there was no case of conduction block or dispersion in IR-CAP. In CIDP, conduction block was observed in 19%–32% of tests in the four nerves and temporal dispersion in 18%–36% of tests in the four nerves. Mean F-wave latencies were below 150% (demyelination marker) of means both in IR-CAP and CIDP. Mean -F-wave increases beyond normal means were 29%–37% in CIDP and 18%–22% in IR-CAP. However, the absence of F-wave was commoner (20% of tests) in CIDP versus 14% of tests in IR-CAP.
Discussion
Chroinc inflammatory axonal polyneuropathy
Our study showed that there is chronic axonal polyneuropathy responding to immunotherapy. Because of lack of a better term, we termed this entity ‘IR-CAP’ for the sake of analysis of data. We believe that this entity is inflammatory in nature in view of the presence of ‘inflammatory cells’ as a prominent finding in the sural nerve biopsy. In 45% of sural nerve biopsies, epineurial inflammatory cells were detected. Axonal neuropathy was another prominent feature in the nerve biopsy, being observed in 51% of sural nerve biopsy. On the basis of these observations, we believe that IR-CAP is best described as CIAP.
Clinical difference between CIAP and CIDP
The clinical profiles of patients with CIAP were quite similar to those of patients with CIDP.8 The clinical characteristics of CIDP (previous episode, high relapse rate and lower rates of antecedent infection, cranial nerve involvement, respiratory failure and spontaneous improvement) are also observed in CIAP.11 12 Relapse of neuropathy is described as the characteristic feature of CIDP from the beginning and is now well regarded as the best clinical index suggestive of CIDP.12 In our study, we had five patients with history of neuropathy. Thus, we consider these findings are strongly suggestive of CIAP, even though it is rare. In our series, one of these three (previous history, antecedent event, and cranial nerve involvement) was observed in 13 patients and immunotherapy was effective in all 13 patients. Proximal muscle weakness is strongly suggestive of polyradiculopathy and CIDP.13–15 Proximal weakness was observed in 76% of IR-CAP cases in contrast to 67% in CIDP. However, one has to rule out other causes such as diabetes. In diabetic amyotrophy, the proximal muscle weakness is one of characteristic findings.16 One significant difference is noted in number of motor neuropathy subtype between CIAP and CIDP, being significantly less common in IR-CAP (p=0.0057).
High CSF protein is similar in mean value and roughly in two-third of patients in both entities. Positive serum autoantibody was observed in two IR-CAP cases. Positive serum autoantibody rate is reported to be low also in CIDP.12
Therapeutically, CIAP is similar to CIDP to the immunotherapy. Though there is less number of cases of ‘improvement to normal’ and more cases of ‘no improvement’ in IR-CAP, suggesting that IR-CAP is a little more resistant to immunotherapy than CIDP, the statistical difference is not significant. Thus, one can conclude that the response to the various immunotherapies is similar between two entities.
Electrophysiological differences between CIAP and CIDP
By the diagnostic criteria, there was no finding of demyelination in CIAP. Motor NCVs and terminal latencies comparison clearly showed that their values are within ‘axonal range’ by the UAB criteria as well as Joint Task Force of EFNS/PNS Guideline.3 10 Thus, the electrophysiological findings are also supported by axonal degeneration in the nerve biopsy.
The NNN-sensory nerve conduction test and triple stimulation test (TST), other sophisticated tests for detection of demyelination, were not done systematically in our patients with IR-CAP to rule out CIDP.17 18 We have done the NNN conduction only in three IR-CAP cases in which axonal neuropathy is found.
Inflammatory nature of CIAP
Inflammatory nature of ‘axonal CIDP’ was suspected clinically in the reported series. However, this was not proven by seven nerve biopsies in the literatures.19–24 It showed axonal degeneration in five but no inflammatory cells. Inflammatory nature of CIAP was pathologically confirmed in one autopsy case so far as described below.25
The most remarkable finding in the present study is demonstration of inflammatory cell and axonal degeneration as the prominent findings in the nerve biopsy in IR-CAP. In contrast, the nerve biopsy in CIDP shows epineurial inflammatory cells in 27%–54% of biopsied nerve and endoneurial inflammatory in 11%–65% of biopsied nerve.7 12 By definition, demyelination, the primary pathological hallmark of CIDP, was not observed in our cases of CIAP. Axonal degeneration is the most common finding in the nerve biopsy, as expected in view of axonal neuropathy pattern in the NCS.
Inflammatory axonal neuropathy is classically seen in vasculitic neuropathy.7 26 We ruled out vasculitic neuropathy by the serial cuts in the nerve biopsy and other clinical and laboratory features in our cases.
In our review of 53 cases with inflammatory axonal neuropathy in the sural nerve biopsy in our Muscle Nerve Histopathology Laboratory, the most common causes were immune-mediated neuropathy in 81% of cases including vasculitis, diabetes mellitus and monoclonal gammopathy as three most common causes.27 This is the reason why vasculitic or diabetic neuropathy was excluded as an evidence of ‘inflammation’ in the nerve biopsy in the third criterion of CIAP.
CIAP versus axonal CIDP
Another possible alternative nosological designation of IR-CAP is axonal CIDP.2 We prefer CIAP, a descriptive term, because axonal CIDP assumes that ‘axonal change in CIAP’ is secondary to CIDP. However, this has not been proven as yet even though the clinical profile and immunotherapy are similar in IR-CAP and CIDP. Sophiscated test such as NNN sensory nerve conduction or TST to rule out demyelination was not done systematically in IR-CAP cases.17 18
Review of 14 CIAP cases in the literature
Pollard et al raised the question on axonal CIDP in 1983 by reporting two cases with chronic relapsing polyradiculoneuropathy, in which nerve biopsy showed axonal degeneration and plasma exchange was not effective in contrast to three cases in which nerve biopsy showed demyelination and plasma exchange was effective.19 Since then, 16 fully described cases of axonal CIDP were reported according to PubMed search.19–25 28–30 Two cases were excluded because of electrodiagnostic feature of demyelination: 25 m/s in motor NCV and 8 ms in latency in median nerve and demyelination in the nerve biopsy in Julien’s case and conduction block in Kataji’s case.22 30 Thus, 14 cases are analysed here. Fourteen cases were all adults >17 years of age and mostly males (six females). All of them had 2 months to 5 years history of symptoms. One case had history of GBS before the relapsing neuropathy. Seven cases had motor neuropathy while six had motor-sensory neuropathy and one, sensory neuropathy. CSF showed a high protein in 10 of 12 tested cases and an increased cell in 2. GM1 antibody was negative in five tested cases. Nerve biopsy in seven cases showed axonal degeneration in five cases and no inflammatory cells. Immunotherapy showed an improvement in 10 of 13 treated cases: complete recovery in 4 cases. Six cases had relapse during the follow-up period. Thus, by definition, 10 cases of reported axonal CIDP had IR-CAP.
Autopsy showed severe axonal loss in the anterior roots and perineurial perivascular lymphocytic infiltration in the mixed nerves and spinal roots in Gordon case and mild cell loss in the dorsal root ganglia but no inflammatory cell or demyelination in the nerve root in Julien’s case.22 25 Julien’s case had an autopsy a year after the neuropathy episode.22 Thus, at least in one case ‘inflammatory polyradiculopathy’ was documented.25
Proposed diagnostic criteria of CIAP
As for the diagnostic criteria of CIAP, we propose three mandatory criteria as described in box 2, all of which were met in our 33 IR-CAP cases. Thus, these can be used as the guidelines for immunotherapy in patients with CIAP.
Proposed diagnostic criteria of chronic inflammatory axonal neuropathy
The diagnosis of chronic inflammatory axonal polyneuropathy was made when all three of the following mandatory criteria are met:
Acquired, chronic progressive or relapsing symmetrical or asymmetrical polyneuropathy with duration of progression >2 months.
Electrophysiological evidence of axonal neuropathy in at least two nerves without any evidence of ‘strict criteria of demyelination’ (box 1).
One of two supportive criteria: a or b.
High CSF protein (>55 mg/dL) or
Inflammatory axonal neuropathy in the nerve biopsy excluding diabetic or vasculitic neuropathy.
The first criterion refers to the clinical feature of neuropathy progressing with duration longer than 2 months. This is identical with the clinical feature in the 1991 ANA and the 2010 EFNS/PNS criteria of CIDP.10 31 This excludes clearly GBS with duration shorter than 4 weeks and subacute inflammatory demyelinating polyneuropathy with duration of 4–8 weeks.8
The second criterion is the major difference between CIAP and CIDP: electrophysiological evidence of axonal neuropathy defined as abnormal nerve conduction without any evidence of demyelination in any nerve conduction. We have adopted the UAB criteria of demyelination, which are based on ‘mean values’ rather than ‘lower limit’ in other systems.3 We prefer mean values because they are well established and clearly published. In contrast, lower limits are not well defined and clearly published. Regardless of lower limit, all of our cases did not meet any criterion of demyelination by Joint Task Force of the EFNS/PNS Guideline.10
The third criterion represents ‘inflammatory nature’ of neuropathy. ‘Inflammation’ is documented by high CSF protein or inflammatory cells in the nerve biopsy. We took this view that the high CSF protein represents inflammatory polyradiculopathy as seen in GBS and CIDP.11 12 Inflammatory cells were documented by the frozen and paraffin sections of biopsied nerve.7 To meet the third criterion of the diagnostic criteria of CIAP, eight cases needed a nerve biopsy in the present study: no CSF test in one and normal CSF protein in seven. Even though the nerve biopsy is invasive, we believe that it is indicated in these cases in view of potential response to immunotherapy in CIAP. We excluded other known causes for inflammatory axonal neuropathy, including diabetic and vasculitic neuropathy, from the third criterion.16 26 27
If one does not have any access to the nerve biopsy, we suggest that one can use any clinical and CSF finding suggestive of ‘CIDP’ and associated gammopathy as the practice guidelines for immunotherapy. These include: previous episode of neuropathy, antecedent events, cranial nerve involvement, respiratory failure, proximal muscles weakness, high CSF protein and gammopathy. If we use these practice guidelines for immunotherapy, then an immunotherapy would have been offered to all patients with IR-CAP except one in the present study. Response to immunotherapy in neuropathy associated with gammopathy has been well known.32
Conclusions
Our study confirmed that CIAP exists and most likely is inflammatory in nature and is similar with CIDP in clinical features and immunotherapy. Major differences between two entities are ‘inflammatory axonal neuropathy’ in nerve biopsy and nerve conduction findings. We prefer CIAP, a descriptive nosological term, because axonal CIDP is not proven as yet. We believe that the recognition of CIAP is important in view of potential therapeutic benefit with immunotherapies. Diagnosis of CIAP can be made by the proposed diagnostic criteria.
References
Footnotes
Original reference Presented at 2005 American Academy of Neurology Meeting. Lu L, Claussen GC, Morgan MB, Oh SJ. Chronic inflammatory axonalpolyneuropathy: A CIDP variant or a distinct entity? Neurology; 64(Suppl 1):A378, 2005.
Twitter @shinjoh@charter.net
Correction notice This article has been corrected since it appeared Online First. References 6, 7, 9, 26, 27, author initials have been corrected to Oh SJ.
Contributors Every one of authors contributed patients during their tenure at the University of Alabama at Birmingham and participated in formulating the this article.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study is approved by the University of Alabama at Birmingham IRB (190425).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Summarised data are available from the first author upon reasonable request.