Article Text

Abstract
Loss of nigrostriatal dopaminergic projection neurons is a key pathology in Parkinson’s disease, leading to abnormal function of basal ganglia motor circuits and the accompanying characteristic motor features. A number of intraparenchymally delivered gene therapies designed to modify underlying disease and/or improve clinical symptoms have shown promise in preclinical studies and subsequently were evaluated in clinical trials. Here we review the challenges with surgical delivery of gene therapy vectors that limited therapeutic outcomes in these trials, particularly the lack of real-time monitoring of vector administration. These challenges have recently been addressed during the evolution of novel techniques for vector delivery that include the use of intraoperative MRI. The preclinical development of these techniques are described in relation to recent clinical translation in an adeno-associated virus serotype 2-mediated human aromatic L-amino acid decarboxylase gene therapy development programme. This new paradigm allows visualisation of the accuracy and adequacy of viral vector delivery within target structures, enabling intertrial modifications in surgical approaches, cannula design, vector volumes and dosing. The rapid, data-driven evolution of these procedures is unique and has led to improved vector delivery.
- neurosurgery
- Parkinson's disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
A major factor contributing to the motor symptoms of Parkinson’s disease (PD) is the loss of dopaminergic projections from the substantia nigra to the striatum resulting in abnormal neuronal and network activity in the basal ganglia–thalamocortical circuit.1 Although PD pathology is known to be complex,2 the idea of counteracting or reversing pathology in the nigrostriatal dopamine circuit has been a focus of the translational neuroscience field since the early 1980s. The first neurosurgical biological clinical trials in PD were focused on replacing the dopaminergic cells that degenerate in PD using cell transplantation techniques.3 However, the challenges of cell acquisition, expansion, survival, circuit integration, function and complications such as graft-induced dyskinesias have helped foster the concept of replacing lost function with genes rather than cells.4 Subsequently, a range of intraparenchymally delivered gene therapies, surgically targeted directly into the basal ganglia, have shown promise in preclinical work. Such biological approaches have potential advantages over the current gold standard surgical treatment, deep brain stimulation (DBS), which is hardware-based, prone to complications such as infection and requires ongoing device programming and battery replacements. Yet, efficacy in phase II gene therapy clinical trials has been limited, in great part due to methodological issues associated with the appropriate surgical delivery of the viral vector.5
One major challenge has been the lack of real-time visualisation of vector delivery, without which it is difficult to improve the distribution of vector during infusions. For this reason, recent clinical trials have employed novel intraoperative monitoring techniques that extend from nearly two decades of preclinical work in non-human primates (NHPs).6–8 In particular, the ability to monitor vector distribution in real time using intraoperative MRI (iMRI) has enabled optimisation of surgical delivery in a recent PD gene therapy clinical development programme, allowing for greater volumes of infusate and improved efficiency of target coverage. Here we review the evolution of this vector delivery method, tracing its transition from preclinical development through ongoing modification in a clinical trial setting.
Initial preclinical development of gene therapy methodologies
Studies in NHPs have been critical for planning safe and appropriate parameters for gene therapy clinical trials, as no other animal allows as close an approximation to vector delivery in the human brain. Preclinical studies of two experimental PD gene therapies in NHPs have demonstrated the importance of adequate tissue coverage and vector dose for improving parkinsonian motor symptoms.7–9 The enzyme aromatic L-amino acid decarboxylase (AADC, also known as dihydroxyphenylalanine (DOPA) decarboxylase) catalyses conversion of levodopa to dopamine, and loss of AADC expression accompanies loss of dopaminergic neurons in PD.10 Preclinical testing of gene therapy using adeno-associated virus serotype 2 (AAV2)-mediated delivery of human AADC (AAV2–human aromatic L-amino acid decarboxylase (hAADC)) began in 1998, as a prodrug approach to express the AADC enzyme in the putamen for localised conversion of levodopa to dopamine. This therapy subsequently underwent extensive evaluation in NHPs with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Early studies established baseline therapeutic responses. In one study, AAV2–hAADC vector was delivered unilaterally to the striatum of parkinsonian animals using the convection-enhanced delivery (CED) method6 11 12 in a total volume of 180 µL per striatum over six injection sites (3.6×1011 vector genome (vg) total dose per animal), resulting in 35%–40% coverage of the striatal target, as assessed by immunohistochemistry.9 The proportion of striatal neurons that expressed the AADC transgene was 5%–6%, which was sufficient to elicit improved behavioural responses to levodopa and increased AADC enzyme activity (assessed by 6-[18F]fluoro-L-meta-tyrosine (FMT) positron emission tomography (PET)) through 8 years of follow-up. In a second, a dose-ranging study in MPTP-treated NHPs, vector doses in the range of 6×1010 vg to 5×1011 vg (total infusion volume 200 µL for all doses) led to increased AADC enzyme activity and improved behavioural sensitivity to levodopa, whereas doses below ≈6×1010 vg had little effect on either measure.7
Preclinical studies involving AAV2–neurturin gene therapy also showed the impact of vector dose. Neurturin is a naturally occurring neurotrophic factor that is a structural and functional analogue of glial cell line-derived neurotrophic factor (GDNF) and plays a role in the survival of nigrostriatal dopaminergic neurons in animal models of PD.13 AAV2–neurturin gene therapy was developed to improve function of degenerating nigrostriatal neurons in PD and provide protection against further neuronal loss. In studies of AAV2–neurturin in NHPs, enhanced expression of tyrosine hydroxylase (TH, the enzyme that catalyses synthesis of the dopamine precursor levodopa from tyrosine) and increased activity in cell survival signalling pathways were observed in a manner dependent on the dose of AAV2–neurturin vector, with effects plateauing above 2×1011 vg.8 These studies further established the importance of infusion volumes that achieve sufficient tissue coverage, using sufficient vector doses, to achieve therapeutic efficacy.
Early studies of experimental gene therapy in animal models used delivery of infusates via manual operation of a microsyringe, leading to limited and focally concentrated coverage within target structures.14 Manual delivery also did not allow for infusing at a consistent flow rate, making measurement of delivery rates inaccurate and thereby introducing unknown variability into the gene therapy administration procedure. CED, on the other hand, uses a pressure gradient generated by an automated pump to distribute high concentrations of macromolecules over significant volumes within the brain parenchyma by fluid convection, allowing for measurable, consistent and predictable infusion rates. This technique was initially designed in animal models to overcome poor passive diffusion of macromolecules through the brain interstitium11 and evolved extensively for the delivery of AAV gene therapies. Intraputaminal CED-mediated delivery of AAV2–hAADC to NHPs with unilateral MPTP-induced parkinsonism was first shown to increase putaminal AADC expression (measured by immunohistochemistry) and enzyme activity (measured by FMT PET).6 Subsequently, CED was used to deliver AAV2 vector containing the GDNF gene (AAV2–GDNF) into the putamen of parkinsonian NHPs, producing high expression levels of GDNF protein.12
Features of the design of the infusion cannula for CED can profoundly impact successful delivery of gene therapy. The initial investigation of AAV2–hAADC delivery in NHPs was carried out using a short fused-silica catheter attached to a syringe pump by Teflon tubing.6 Two issues arose with this device assembly: the tubing had a considerable internal ‘dead’ volume, which was filled with oil to conserve vector volume, and a large quantity of vector was lost to adsorption in the Teflon tubing itself. To address these issues, Teflon tubing and stainless steel cannulas were lined with silica and the internal volume was reduced. Additionally, the length of each infusion cannula was measured to ensure that the distal tip extended approximately 3–4 mm beyond the length of the respective guide, to create a stepped design at the tip of the cannula. Combined with increased infusion flow rates, these modifications resulted in greatly improved target coverage when evaluated in NHPs.15 However, higher flow rates were associated with backflow of infusate up and around the infusion cannula, which resulted in delivery of therapeutic agent outside of the intended target. To avoid backflow, the stepped-design concept was incorporated directly into the creation of a new cannula. This cannula design resulted in backflow-free delivery at increased flow rates during delivery of CED-infused liposomes to multiple brain regions in NHPs.16 Further evolution of this cannula design occurred at the same time as development of real-time MRI monitoring, but at the time of these developments, most clinical trials in PD were not even employing CED or step cannulas, as will be discussed in the next section.
Initial clinical testing of experimental gene therapies for PD
The first experimental gene therapy to undergo clinical evaluation in PD was AAV2-mediated delivery of glutamic acid decarboxylase (GAD, the enzyme that synthesises the inhibitory neurotransmitter γ-aminobutyric acid (GABA)) to the subthalamic nucleus (STN). The objective of AAV2–GAD gene therapy was to reduce pathological overactivity in the STN by promoting local GABA synthesis. In the initial open-label phase I trial, initiated in August 2003, patients had significantly improved Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores on and off medication at 3, 6 and 12 months versus baseline.17 In the randomised, placebo-surgery controlled phase II trial, UPDRS motor scores showed a significantly greater improvement in patients who received AAV2–GAD versus those who underwent placebo surgery at 1, 3, 6 and 12 months of follow-up.18 19 Although both the phase I and phase II trials used CED for delivery, the vector dose, volume and infusion rate were (1) not validated in prior preclinical NHP studies and (2) lower in the phase II trial than in all of the subsequent gene therapy trials (online supplementary table 1). These trials involved conventional surgical techniques using a stereotactic frame to deliver the infusion cannula and ‘blind’ infusions. In the absence of intraoperative monitoring, it was not possible to determine how these factors affected coverage of the STN.
Supplemental material
AAV2–hAADC experimental gene therapy was designed to provide AADC enzymatic activity in the putamen for potential localised conversion of levodopa to dopamine. Two open-label phase I trials tested this approach in the mid-2000s—one in the USA and one in Japan—in which AAV2–hAADC was delivered at doses of up to 1.5×1011 vg in 100 µL infusion volumes to each putamen. The US trial was the first to employ the stepped-design cannula, which had undergone testing in NHPs, using CED for vector delivery. Significant improvements in on-medication and off-medication total UPDRS scores were observed through 12 months in the US trial, but outcomes deteriorated thereafter.20 21 In the trial conducted in Japan that mirrored the design of the US trial, modest benefits in motor symptoms were observed through the last follow-up at 6 months.22 Low vector dosing as well as insufficient infusion volumes (leading to insufficient target coverage) likely played a role in the limited efficacy of AAV2–hAADC in these clinical trials. The authors of the US trial acknowledged that higher dosing and infusion volumes would be a consideration for future trials, with the aim of achieving greater coverage of the putamen.21 However, reliable determination of target coverage in vivo requires real-time intraoperative monitoring of vector infusion, which was not available at the time of these trials.
A novel enzyme replacement strategy used a different viral vector, carrying genes for not one but three enzymes involved in the dopaminergic pathway. The enzymes TH and cyclohydrolase 1 (CH1) are required for conversion of the amino acid tyrosine to levodopa, which is the precursor for AADC-mediated dopamine synthesis. Lentiviral vector-mediated delivery of TH, AADC and CH1 (lenti-TH/AADC/CH1) to the putamen, thought to provide the enzymatic machinery necessary for local continuous dopamine production, was evaluated in a phase I/II clinical trial.23 24 In this study, lenti-TH/AADC/CH1 was administered manually via a hand-operated syringe across three increasing dose levels using conventional stereotactic techniques, with reduced-diameter cannulas (designed to provide greater flow rates and enhanced vector distribution) used in later dose-level cohorts. Infusion volumes were not provided in the published trial results. Modest improvements in off-medication UPDRS motor scores at 12 months versus baseline were observed in the 15 patients administered lenti-TH/AADC/CH1 at different dose levels, but there were no cohort-dependent effects, despite increased dosing and changes to infusion techniques. The use of manual delivery (vs CED) precluded accurate measurement of infusion flow rates, and without intraoperative monitoring, it was not possible to confirm whether the altered infusion methods had any effect on vector distribution. Further, efficacy was difficult to meaningfully evaluate as there was no placebo-surgery control group, and the authors acknowledged that the magnitude of effects was in the range of placebo effects observed in the other gene therapy trials.23 24 Clinical evaluation of a lenti-TH/AADC/CH1 vector that is more potent in promoting dopamine synthesis in vitro25 is currently under way.
As discussed previously, delivery of the neurotrophic factor neurturin to basal ganglia structures, using AAV2, is an alternative to enzyme replacement gene therapy approaches in PD. Initial open-label phase I and placebo-surgery controlled phase II trials investigated bilateral putaminal administration of AAV2–neurturin using conventional stereotactic frames and blind infusions with manual operation of infusion syringes. Vector doses similar to those in the initial AAV2–hAADC trials but with low infusion volumes (40 µL per putamen) were given (online supplementary table 1).26 27 In the first phase II trial, there was no significant difference in the effect on the UPDRS III motor score, the primary endpoint, between the AAV2–neurturin-treated and the placebo-surgery groups.26 The authors suggested that this was at least partly due to a failure of retrograde transport of neurturin from the putamen to substantia nigra. Subsequent preclinical work, however, suggested that although the degenerating nigrostriatial pathway might not provide an adequate conduit for retrograde transport of neurotrophic factors, anterograde transport between the putamen and substantia nigra is maintained.28 29 Insufficient infusion volume and target coverage and a lack of availability of intraoperative infusion monitoring at the time likely played a significant role in the observed lack of clinical efficacy. In two subsequent phase I and phase II trials, AAV2–neuturin was administered to both putamen and substantia nigra using CED rather than manual infusion, with greater vector doses and infusion volumes for putaminal administration (150 µL per putamen).30 31 Nevertheless, the phase II trial did not demonstrate significant improvement in motor symptoms as compared with placebo surgery.30 31 Postmortem staining for neurturin protein in four patients showed that, at best, 15%–22% of the targeted putaminal volume had been achieved,32 dramatically underscoring the potential value of using real-time visualisation of vector delivery to confirm adequate target coverage.
Development and validation of iMRI-guided CED in NHPs
The need to maximise on-target while minimising off-target vector distribution has propelled the development of real-time MRI guidance of vector delivery. To achieve this goal, the neurosurgeon must be able to see the infusate distribute in the brain and act on this visual information to modify the ongoing infusion in real time. The iMRI monitoring of CED is accomplished by admixing a contrast agent such as gadoteridol with the gene therapy vector to permit visualisation of the combined infusate in near real time during administration, with serial MRI scans obtained every 2–4 min. Following studies in rodent models,33 iMRI monitoring was used in NHPs to successfully visualise distribution of gadoteridol-containing liposomes delivered by CED to a variety of brain regions.34 Subsequent NHP studies demonstrated that iMRI monitoring could provide real-time detection of leakage of these liposomes outside the target structure as well as cannula track backflow.35 In addition, iMRI monitoring demonstrated the critical and previously underappreciated role of the perivascular spaces in off-target drug distribution.36 37 Subsequently, iMRI-guided CED was validated for tracking AAV2 vector distribution in NHPs. Importantly, distribution of gadoteridol during coinfusion with both AAV2–hAADC and AAV2–GDNF into the thalamus was found to closely match gene expression patterns.38 39 These studies demonstrated that, with these particular therapeutic agents, visualisation of gadoteridol enhancement within the brain is a reasonable surrogate for the location of subsequent gene expression.
Another important step in improving the targeting of brain-delivered gene therapy has been the development of an MRI-compatible targeting system. The ClearPoint® system (ClearPoint Neuro (formerly MRI Interventions), Irvine, California, USA) was adapted for vector infusions in NHPs. This system, which consists of an MRI-compatible skull-mounted aiming device (SmartFrame®) and MRI-integrated software, was developed for stereotactic placement of devices into the brain and has become a widely used platform for iMRI-guided implantation of DBS electrodes and other devices.40–43 When adapted for vector delivery, a ceramic, fused silica cannula (SmartFlow®, ClearPoint Neuro) is used.5 The first reported use of the ClearPoint platform for targeted infusion was in NHPs, providing the initial demonstration of submillimetric accuracy and uniform distribution of infusate from the delivery cannula.44 The precision, predictability and safety of this system were validated across multiple brain targets in NHPs, with mean targeting errors of <1 mm and linear relationships observed between gadoteridol infusion volumes and distribution volumes. No haemorrhages or evidence of tissue damage were observed, with minimal inflammation.44 In a follow-up study intended to estimate infusion parameters for a clinical trial of AAV2 vector delivery to the putamen, the colocalisation of contrast agent and gene expression was again confirmed using the clinical platform.38 39 It should be noted that by this point, all iMRI-CED in NHPs had been performed through a frontal approach, similar to that used for DBS lead implantation.
Initial use of iMRI-guided CED in a gene therapy clinical trial
The iMRI-guided CED for gene therapy was first employed in a recently reported phase I open-label dose-escalation trial of AAV2–GDNF administration to the putamen of patients with advanced PD. Similar to AAV2–neurturin, this experimental gene therapy was designed to provide GDNF-mediated neurotrophic support to nigrostriatal neurons.45 Patients were administered AAV2–GDNF at doses of 9×1010 to 9×1011 vg in 450 µL infusate volumes per putamen using a frontal approach and ClearPoint. In this small safety trial (n=13), stabilisation of UPDRS scores (through 18 months of follow-up) and medication requirements (48 months) were observed. Real-time tracking of coinfused gadoteridol and post hoc analysis revealed that putaminal coverage was low (mean 26%), which was thought to be partly due to the infusate distributing along perivascular spaces and extending outside of the putamen.45 Enrolment was closed prematurely, due in part to the limited putaminal coverage, which did not meet the 35% threshold needed for efficacy as suggested by preclinical studies.9 This course of events suggests that the investigators may not have fully used the opportunities provided by real-time visualisation to acutely modify infusions or to modulate the infusion strategy over time.
Employment of iMRI-CED in the VY-AADC clinical development programme
Implementation of iMRI-monitored CED in the recent VY-AADC (AAV2–hAADC) clinical programme has resulted in a relatively rapid improvement in vector delivery technique, enabled by a proactive response to real-time visual information during each infusion. In this programme, our ability to monitor vector distribution during surgery resulted in reactive and then proactive manoeuvres to introduce significantly larger vector volumes and better target coverage than had been achieved in earlier trials. Nonetheless, several challenges associated with surgical and vector infusion techniques were revealed.
iMRI monitoring informed within-trial technical and surgical changes: PD-1101 trial
PD-1101 (NCT01973543) is a phase I, open-label, dose-escalation safety trial of VY-AADC01, an investigational gene therapy consisting of AAV2 vector carrying the human AADC gene, in patients with advanced PD and medically refractory motor fluctuations. This programme was initiated at University of California, San Francisco, and was the first PD clinical trial to report the use of iMRI-CED for the delivery of an experimental gene therapy.46 Patients received bilateral, simultaneous putaminal infusions of VY-AADC01 at one of three vector-genome doses, delivered using skull-mounted SmartFrames, the ClearPoint neuronavigational system and SmartFlow cannulas. VY-AADC01 was admixed with gadoteridol, allowing for continuous, serial imaging to monitor infusate distribution and target coverage. The surgical goal was to cover as much of the postcommissural putamen as possible, given that it is the sensorimotor portion of the putamen47 and shows greater loss of dopaminergic terminals in PD.48 At least two trajectories per putamen were necessary, using a traditional frontal approach, similar to prior gene therapy trials and standard DBS lead implantations.
The iMRI monitoring during VY-AADC01 administration revealed perivascular leakage of infusate into adjacent structures, and backflow up the cannula tracks associated with the infusion procedure (figure 1A,B and online supplementary video 1A,B). To address the issue of perivascular leakage, several modifications were made to the infusion technique. Single-point infusions were used in the first three patients in the PD-1101 trial, with the cannula tip positioned in the centre of the putamen. This initial strategy was based on the assumption that CED delivery in the human would result in a relatively evenly distributed sphere around the distal portion of the cannula. However, real-time MRI monitoring of the infusions revealed that perivascular leakage was often more significant than anticipated. The only way to overcome this off-target delivery was to advance the cannula deeper into the tissue during infusion in order to move its tip past the offending perivascular space. For the next four patients treated, stacked infusions were used, such that infusions were made at multiple (usually 2) points, with the proximal point infused first, followed by cannula advancement and infusion at the distal point (figure 1C and online supplementary video 1C). It should be noted that distal infusion followed by proximal withdrawal of the cannula was not attempted, as this would create a larger potential space between the cannula and tissue, potentially leading to backflow. Finally, for the last eight patients, a technique of progressive stepped advancement during the entire infusion from proximal to distal was adopted based on NHP experience49 (figure 1D and online supplementary video 1D). This technique was found to increase the surgeon’s ability to achieve infusions that ‘filled’ the putamen as uniformly as possible while providing better ability to move the cannula tip away from problematic perivascular spaces.
Supplementary video
Supplementary video
Supplementary video
Supplementary video

Intraoperative monitoring of VY-AADC01 admixed with gadoteridol. Backflow up cannula track (A) and perivascular and off-target leakage (B) in PD-1101. Stacked infusions (C) and progressive cannula advancement (D). Posterior approach with progressive cannula advancement employed in PD-1102 (E).
Real-time MRI monitoring of the infusions also revealed that distribution beyond the superior boundary of the putamen was more common than realised. This was particularly true if there was excessive side-to-side movement of the cannula during insertion or uneven movement of the MRI table as the patient was being moved to the centre of the scanner for imaging. To address backflow beyond the superior aspect of the putamen, modifications were made to the cannula design. All cannulas used in the PD-1101 trial were constructed with two steps in outer diameter. The initial design had a small, square step 3 mm from the cannula tip and a second, bullet-shaped step 18 mm from the cannula tip. Both steps were known to resist backflow, but the larger second step was particularly effective at keeping infusate in the target once inserted into the putamen. However, when using a frontal trajectory, the 18 mm distance from the cannula tip to the second step meant that, by design, the second step would not enter the putamen unless the tip was deep in the ventral region of the putamen and in close proximity to the lenticulostriate arteries. Cannulas with different step geometries were manufactured, and a design with a shorter distance to the second step (13 mm) was selected so that the second step would cross the dorsal boundary of the putamen earlier and restrict backflow outside of the putamen (figure 2). This updated design was used for patients in cohorts 2 and 3 (patients 6–15) of PD-1101.
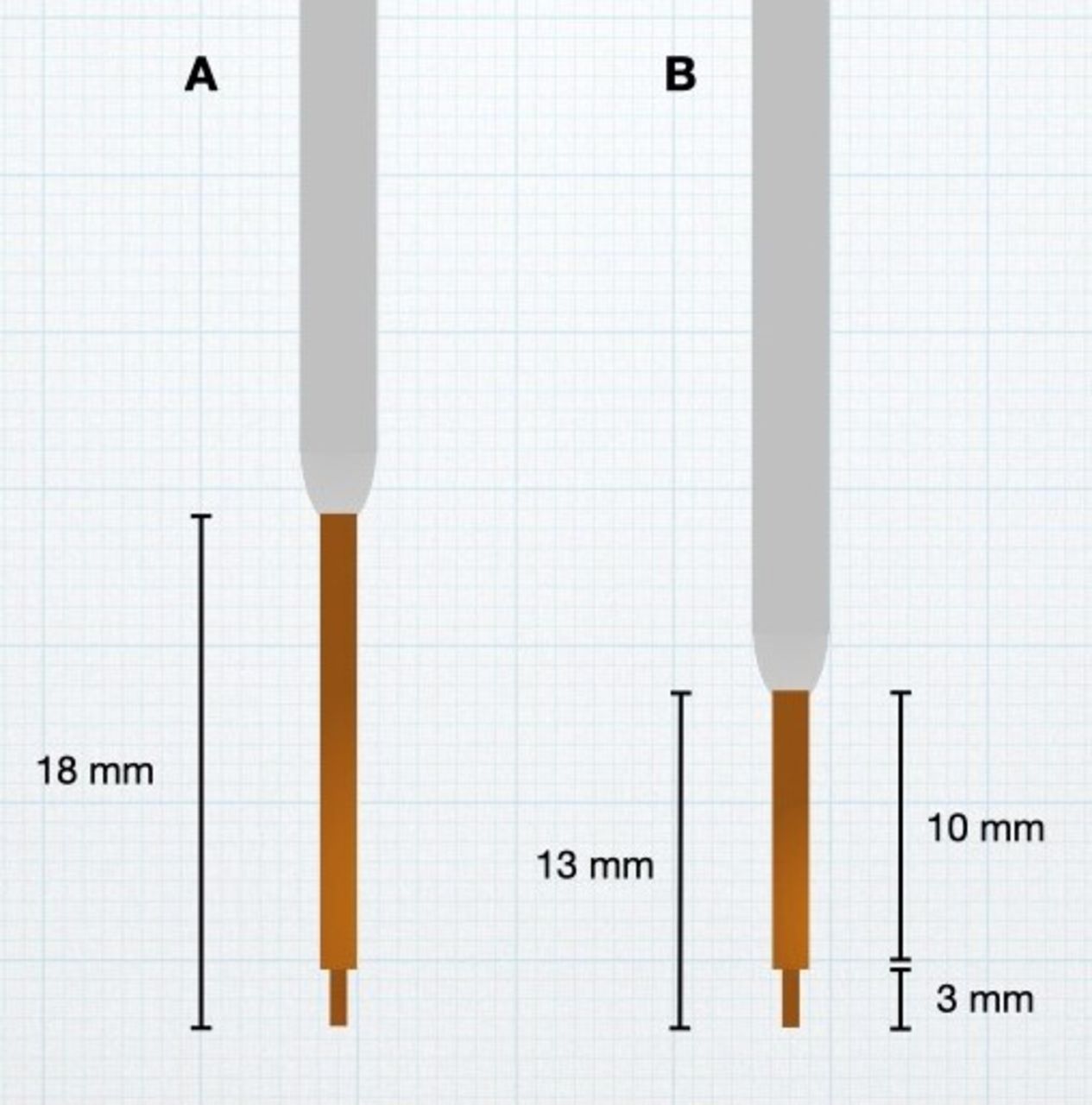
Stepped-design cannulas. Cannulas with two-diameter steps were employed in PD-1101, with the second step known to resist backflow past the first step after target insertion. The cannula tip was shortened from 18 mm (A) to 13 mm (B) for use in cohorts 2 and 3 of PD-1101 and PD-1102.
In the original US and Japanese trials of AAV2–hAADC, patients were administered 100 µL of vector infusate split between two infusions (50 µL each) per putamen, providing a maximum dose of 3.0×1011 vg.20 22 In the first cohort of PD-1101, real-time iMRI monitoring allowed the safe administration of up to 450 µL of infusate per putamen at a concentration of 8.3×1011 vg/mL, providing a total dose of up to 7.5×1011 vg and similar to the infusion parameters employed in the AAV2–GDNF trial described previously.45 Despite this significant increase in volume over the initial AAV2–hAADC trials, the mean total putaminal coverage in this cohort was 21% (20% postcommissural) (figure 3), similar to the AAV2–GDNF trial and again well below the 35% threshold suggested to be necessary for efficacy in preclinical NHP studies.9 The volume of infusate was doubled to 900 µL per putamen for cohorts 2 (vector concentration unchanged) and 3 (vector concentration increased to 2.6×1012 vg/mL). These modifications led to an increase in mean total putaminal coverage to 34% (39% postcommissural) in cohort 2 and a further increase to 42% (53% postcommissural) in cohort 3. The improvement in coverage despite consistent volumes between cohorts 2 and 3 is a direct result of incremental improvements in the infusion technique that were implemented in response to real-time MRI feedback as the trial progressed.

Infusion volumes and durations, vector concentrations and putaminal coverage in PD-1101 and PD-1102 trials. vg, vector genome.
Importantly, this increasing coverage of the putamen led to successive increases in AADC enzyme activity, as measured by [18F]-fluoro-L-DOPA (18F-DOPA) PET (figure 4A,B).46 A strong correlation was observed between percentage coverage of the putamen measured by gadoteridol distribution on iMRI and change in AADC activity measured by 18F-DOPA PET at 5–7 months post-VY-AADC01 administration (r=0.84, p=0.0002; shown combined with PD-1102 trial data, discussed below, in figure 4C).50 With regard to clinical outcomes in the PD-1101 trial, daily antiparkinsonian medication requirements were reduced in cohort 2 (through 24 months of follow-up) and cohort 3 (18 months).46 Clinical outcomes were also durably improved, with meaningful reductions in on-medication (cohorts 2 and 3) and off-medication (all cohorts) UPDRS III scores that persisted through last follow-up (36, 24 and 12 months after VY-AADC01 administration in each cohort, respectively). Increased diary good quality ON times (ON time with no dyskinesia plus ON time with nontroublesome dyskinesia) were reported by patients in all cohorts.

AADC enzyme activity as measured by 18F-DOPA PET in individual patients from PD-1101 (A), by cohorts (baseline and at 6-month measurements) in PD-1101 and PD-1102 (B), and correlation of the change in 18F-DOPA PET with putaminal coverage measured by MRI (C). PET imaging frames captured 65–75 min after 18F-DOPA administration. Box plot in panel B shows median (middle line), 25th–75th percentiles (box) and range (bars). aSORs were calculated using bilaterally averaged occipital time–activity curve (kBq/mL) region-of-interest values in each patient. bn=7; analysis of scan for one patient in PD-1102 was confounded by movement artefact, and data for this patient were excluded. cData were not available for one patient from PD-1101 cohort 1 and one patient from PD-1102. dTwo overlapping data points from PD-1101 cohort 1 and cohort 2. eTwo overlapping data points from PD-1102. AADC, L-amino acid decarboxylase; 18F-DOPA, [18F]-fluoro-L-dihydroxyphenylalanine; PET, positron emission tomography; SOR-1, striatal-to-occipital ratio-1; SUVR, standardised uptake value ratio; vg, vector genome.
Although the clinical and patient-reported outcomes reported in the open-label, dose-escalation PD-1101 trial showed potential benefit, we hypothesised that further increases in tissue coverage would lead to greater clinical efficacy. In addition, there were several motivations to improve the overall efficiency of the infusion procedure itself. Anatomically, the putamen is elongated in the anterior–posterior direction, tapering significantly in the postcommissural ‘tail’ portion. In addition, the majority of the perivascular spaces in the putamen originate at the base and course superiorly. The frontal approach was initially selected because it is familiar to stereotactic surgeons, is easy to perform with a patient supine in an MRI scanner and generally allows straightforward trajectory planning. However, this approach orients the trajectories close to 90° to the long axis of the putamen, which has several disadvantages. First, at least two trajectories are needed to provide adequate coverage, and surgical procedures in PD-1101 frequently took 8–10 or more hours to complete. Second, the angle of frontal trajectories makes it particularly difficult to achieve the desired coverage in the sensorimotor regions of the putaminal tail. Third, the length of the cannula pass within the putamen is relatively short, which leads to greater potential for off-target delivery above the putamen if backflow occurs at the beginning of the infusion. Finally, if perivascular leakage occurs, advancing the cannula past the offending perivascular space may not be successful as the cannula trajectories are often parallel to the course of the putaminal vasculature.
A posterior trajectory, with an entry point in the parieto-occipital region and a trajectory that enters the putaminal tail and proceeds parallel to the long axis of the structure, was suspected to be more efficient. Experience with the posterior approach was first acquired in NHPs,51 and its safety is further supported by a growing clinical experience with stereotactic laser ablations in the temporal lobe for epilepsy that employ similar trajectories.52 53 Hence, in the second phase I trial of VY-AADC01 (PD-1102), a posterior approach to the putamen was used for the first time, with the goal of achieving 50% coverage of the total putamen and maximising postcommissural coverage using a single cannula pass.
Continued surgical evolution: PD-1102 trial
PD-1102 (NCT03065192) is an ongoing, open-label phase I trial. To further enhance coverage of the posterior putamen and reduce the number of trajectories necessary, a posterior (parieto-occipital) approach using ClearPoint with the patient in the prone position was used for delivery of VY-AADC01 in this second trial. In addition to this change in approach, vector infusion volume was increased again to a maximum of 1800 µL per putamen at a concentration of up to 2.6×1012 vg/mL. Posterior trajectories were found to provide better coverage of the putamen (particularly the tail) and afforded the surgeon greater ability to advance the cannula away from troublesome perivascular spaces. The approach even allowed backflow to the second cannula step to work in the surgeon’s favour. Since the length of the cannula pass within the putamen is much longer than with a frontal approach, the second cannula step was found to enter the putamen much earlier in the infusion process. Once the second step is inside the putamen, the infusion rate can be increased more aggressively, allowing backflow to actually enhance coverage of the putaminal tail (figure 1E and online supplementary video 1E) as shown in our NHP studies.49 51
Supplementary video
The modifications to the surgical procedure made in PD-1102 resulted in total putaminal coverage exceeding the 50% goal (figure 5), with a mean 76% coverage of the postcommissural putamen. The shift to the posterior approach and concomitant reduction in trajectories also reduced infusion durations and overall procedure times (figure 3). AADC enzymatic activity was further increased in PD-1102 compared with PD-1101 (figure 4B), such that putaminal coverage and change in AADC activity remained correlated when incorporating PD-1102 data into the PD-1101 analysis (r=0.87, p<0.0001; figure 4C). Daily antiparkinsonian medication requirements were reduced at 6 and 12 months post-VY-AADC01.54 Of the eight patients who underwent VY-AADC01 administration in PD-1102, two had mild intraoperative intracerebral haemorrhages. In both cases, procedures continued as planned. Postoperatively, one patient was asymptomatic and the other experienced a temporary visual disturbance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship between AAV2 gene therapy infusion volumes and putaminal coverage. Data are based on analysis of iMRI images progressively acquired during putaminal infusions of VY-AADC01 (PD-1101 and PD-1102 trials), AAV2-GDNF, and AAV2-NTRN in patients with PD.45 46 54 Putaminal coverage was measured at infusion volumes matching those used in earlier gene therapy trials, to infer volumes of distribution that would have been provided by those infusion volumes. Values do not represent actual coverage from trials that did not employ iMRI monitoring.20–22 26 27 30 31 55 AADC, aromatic L-amino acid decarboxylase; AAV2, AAV serotype 2; GDNF, glial cell line-derived neurotrophic factor; iMRI, intraoperative MRI; NRTN, neurturin; PD, Parkinson’s disease; Vd, volume of distribution; vg, vector genome; Vi, volume of infusion.
The surgical and infusion techniques evaluated in PD-1102 have informed the design of RESTORE-1 (PD-1105, NCT03562494), a randomised, placebo-surgery controlled, double-blind phase II trial of VY-AADC02 (an identical vector to VY-AADC01 made with updated manufacturing techniques) that is ongoing in patients with PD and refractory motor fluctuations.
Conclusions
Initial experimental gene therapies for PD showed limited efficacy in early clinical trials. These trials used blind infusions without real-time monitoring of vector distribution, preventing investigators from appreciating the extent to which low volumes and off-target delivery led to inadequate target coverage, gene expression and therapeutic benefit. The development of iMRI-guided CED and its use in the VY-AADC clinical development programme has established a new era of direct intraparenchymal drug delivery, in which ongoing modification of both individual procedures and overall clinical trial design is not only possible but also imperative for optimising clinical outcomes. Our modifications—including progressive advancement of cannulas during the infusion, updated cannula design, switching to a single posterior trajectory for the putamen and increased infusion volumes—have significantly improved target coverage and reduced surgical time. The modifications described in this paper are currently being applied to a number of gene therapies under development that use intraparenchymal delivery of agents that codistribute with a contrast agent. This rapid technical evolution during an ongoing neurosurgical clinical trial, informed by both preclinical testing in NHPs and active responses to intraoperative imaging findings, serves as a model for the development of other similar therapeutics. We anticipate that with continued evolution in surgical technique, surgical delivery of gene therapy can be improved. In turn, this will provide a better understanding of the efficacy of a gene therapy approach and its potential advantages relative to other neurosurgical interventions such as DBS.
Acknowledgments
The authors thank the people with Parkinson's disease and their families who participated in these gene therapy trials and the study support teams. We thank Elisabeth M Fine of Voyager Therapeutics, Inc, for her thoughtful review of the manuscript. Medical writing and editorial support were provided by Jeremy Kennard, PhD, and Jason Tuffree of Ashfield Healthcare Communications, Middletown, CT, and were funded by Voyager Therapeutics.
References
Footnotes
Correction notice This article has been corrected since it appeared Online First. Figure 2 has been replaced.
Contributors RMR, APK and PSL contributed to the conception of the review and prepared the initial draft with the assistance of a medical writer. RMR, KSB, CWC, ADVL, REG, RL, SAF, SKK, APK, BR and PSL reviewed the manuscript critically and revised it for intellectual content, approved the final manuscript before submission, and are accountable for the accuracy and integrity of the work.
Funding The Michael J. Fox Foundation and Voyager Therapeutics, Inc, provided funding for the PD-1101 and PD-1102 trials, including for data collection and analyses.
Competing interests RMR, CWC, ADVL, REG and SAF received grants from Voyager Therapeutics, Inc. KSB received grants and personal fees from Voyager Therapeutics. Voyager Therapeutics is funding this research and is developing products related to the research described in this paper. REG serves as a consultant to Voyager Therapeutics and personally receives compensation for these services. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. RL received consulting fees from Voyager Therapeutics. SKK has nothing to disclose. APK is a former employee of Voyager Therapeutics and owns stock in that company. BR is a former employee of Voyager Therapeutics. PSL has received grants from Voyager Therapeutics and non-financial support from ClearPoint Neuro (formerly MRI Interventions, Inc).
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.