Article Text

Abstract
The immune system has long been recognised important in pain regulation through inflammatory cytokine modulation of peripheral nociceptive fibres. Recently, cytokine interactions in brain and spinal cord glia as well as dorsal root ganglia satellite glia have been identified important— in pain modulation. The result of these interactions is central and peripheral sensitisation of nociceptive processing. Additionally, new insights and the term ‘autoimmune pain’ have emerged through discovery of specific IgGs targeting the extracellular domains of antigens at nodal and synaptic structures, causing pain directly without inflammation by enhancing neuronal excitability. Other discovered IgGs heighten pain indirectly by T-cell-mediated inflammation or destruction of targets within the nociceptive pathways. Notable identified IgGs in pain include those against the components of channels and receptors involved in inhibitory or excitatory somatosensory synapses or their pathways: nodal and paranodal proteins (LGI1, CASPR1, CASPR2); glutamate detection (AMPA-R); GABA regulation and release (GAD65, amphiphysin); glycine receptors (GLY-R); water channels (AQP4). These disorders have other neurological manifestations of central/peripheral hyperexcitabability including seizures, encephalopathy, myoclonus, tremor and spasticity, with immunotherapy responsiveness. Other pain disorders, like complex regional pain disorder, have been associated with IgGs against β2-adrenergic receptor, muscarinic-2 receptors, AChR-nicotinic ganglionic α-3 receptors and calcium channels (N and P/Q types), but less consistently with immune treatment response. Here, we outline how the immune system contributes to development and regulation of pain, review specific IgG-mediated pain disorders and summarise recent development in therapy approaches. Biological agents to treat pain (anti-calcitonin gene-related peptide and anti-nerve growth factor) are also discussed.
- autoimmune
- pain
- chronic
Statistics from Altmetric.com
Introduction
The immune system’s involvement in pain was described by Celsus in the first century where redness (rubor), warmth (calor) and swelling (tumour) were recorded as accompanying pain (dolor).1 Pain and its immune-mediated accompaniments are healthy responses to prevent initial injury and accelerate tissue recovery. However, when pain is out of proportion to injury, a pathological process is implied. Severity of pain is determined by complex peripheral and central integration of spinal, limbic and cortical pathways, all with potential immune mediation.2 Psychosocial factors, mood and age can all influence pain perception.3 According to WHO, more than a quarter of the population is suffering chronic pain, defined as pain lasting more than 3 months. Traditional analgesic drugs are often not efficacious in this population. For patients with chronic neuropathic pain, daily scheduled opioids have a major negative impact through increased morbidity, without clear improved functional outcomes.4 Phenotypic classifications based on pain generators form the basis for the current pain classifications.5 Neurologists are involved when pain is thought to be neuropathic and associated with a lesion or disease of the somatosensory nervous system. Guidelines have been created to help determine whether pain is neuropathic.6 Important in that consideration is whether there are (1) sensory loss and (2) sensory gain with hyperalgesia and/or allodynia in a neuranatomically plausible distribution and whether investigations confirm a lesion of the somatosensory nervous system. When no pain generator is found, a somatoform pain disorder is commonly diagnosed.7
The aim of this review is to explore current understanding of the immune system’s role in pain generation, discuss emerging autoimmune neurological pain disorders and highlight important shifts in how we diagnose and treat affected patients. Included in the review is a discussion of the development of new biological immune therapies being considered for common pain disorders not felt to have an autoimmune cause.
Immune-mediated triggers and effectors of pain
Innate immunity and pain
Activation of neurogenic inflammation through innate immunity plays a key role in initiating and maintaining neuropathic pain.8Afferent nociceptive nerves communicate with the immune system to produce integrative healing of injured tissues. Tissue injury causes a release of endogenous danger signals, termed alarmins, which are recognised by toll-like receptors, triggering neurogenic inflammation via nociceptive nerve fibres.8 These fibres are also activated through cytokine receptors, danger receptors and pathogen-associated molecular receptors (figure 1). This response drives an efferent release of neuronal mediators to increase blood flow (vasodilatation), heighten pain sensitivity (through reduced nerve membrane firing thresholds and slowed membrane repolarisation) and stimulate both chemotaxis and T-cell priming at the site of injury. The major neuronal mediators are neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P. These neuropeptides can also induce pain by stimulating proximal effectors to sensitise nociceptors.9 Cytokines are the main effectors of the response to tissue injury with two opposing phenotypes: the pro-inflammatory, IL-1β, TNF, IL-6, IL-15, IL-17, IL-18 and IFN-γ; and anti-inflammatory, IL-4, IL-10 and TGF-β (table 1). In neuropathic pain modulation, cytokines are either predominantly algesic or analgesic. In chronic pain, there is often an imbalance between the major cytokine algesic and analgesic mediators released by activated immune and immune-like glial cells.

Immune mediators and autoimmune targets in pain regulation. The targeted antigens and receptors associated with neurogenic inflammation and autoimmune pain without inflammation are shown throughout the neuraxis; each is important in neural nociceptive hyperexcitability and pain. (Bottom) Peripheral nociceptive pain sensitisation. Peripheral nociceptive sensitisation refers to hyperexcitation of nociceptor fibres by potent or persistent stimuli; hyperexcitation can occur through afferent PAMP receptors (NIRs, IRLs), danger receptors (TRPA1, TRPV1 DAMP) and cytokine receptors (IL-1Βr, TNF-αR). The efferent mediators are subsequently released by neurons, including substance P, calcitonin gene-related peptide, adrenomedullin, cytokines and glutamate, resulting in vessel dilation and lymphocyte, macrophage and neutrophil recruitment. The neural mediator cytokines are diverse and can directly alter pain fibre firing thresholds. (Right) Clonal expansion of T and B cells in autoantibody generation. In immunoglobulin-based disorders, production of autoantibodies against tumours, infectious agents and many unknown antigens through clonal expansion can cause nociceptive hyperexcitability. (Centre) Synaptic CNS and PNS regulators of pain. The CNS ascending and descending nociceptive pathways are important in pain regulation (shown in red) and influenced by multiple synaptic IgG targeted antigens (AQP4, GlyR, GAD, amphyphysin, P/Q and N-type VGCC) in autoimmune disease. Peripheral synaptic autoantibody targets include the muscle AChR, neuronal ganglionic AChR (α3), β2AR and M2R, which are associated with complex regional pain syndrome. (Left) Nodal IgG targets in autoimmune pain. The strongest evidence for autoimmune pain disorders comes from two IgGs discovered to target nodal protein (LGI1 and CASPR2); others also have been described (Contactin-1, CASPR1). AChR, acetylcholine receptor; AQP4, aquaporin-4; CASPR2, contactin-associated protein-2; β2AR, β2-adrenergic receptor; CNS, central nervous system; DAMP, danger-associated molecular pattern; DRG, dorsal root ganglion; GAD, glutamic acid decarboxylase; GlyR, glycine receptor; LGI1, leucine-rich glioma-inactivated 1 protein; M2R, muscarinic-2 receptor; PAMP, pathogen-associated molecular pattern; PNS, peripheral nervous system; VGCC, voltage-gated calcium channel; VGKC, voltage-gated potassium channel.
Predominant cytokine algesic and analgesic neural mediators
Another important factor in pain stimulation are microglia, the resident macrophage cells of the central nervous system (CNS) (figure 2).10 Recent studies demonstrate microglial activation is important in chronic pain modulation.11 Chronic pain can also lead to brain microglia proliferation and activation at the cortex, thalamus, amygdala and hypothalamus, resulting in central pain sensitisation.2 Spinal microglia are activated in the very early phase (within 4 hours) after peripheral nerve injury. The injured afferents are able to release colony stimulating factor-1, which will trigger microglial proliferation in the spinal cord and adenosine triphosphate (ATP), which activates the P2RX4 (purinoreceptor for ATP) expressed on microglia resulting in them adopting a pro-inflammatory phenotype.12–14 Supraspinal microglia can also be activated by peripheral nerve injury and modulate chronic pain. Activated microglial cells are able to release a variety of diffusible factors including pro-inflammatory cytokines, brain-derived neurotrophic factors and proteases. These microglia-derived factors can signal to astrocytes and neurons, which enhance neuronal firing through both direct and indirect mechanisms.
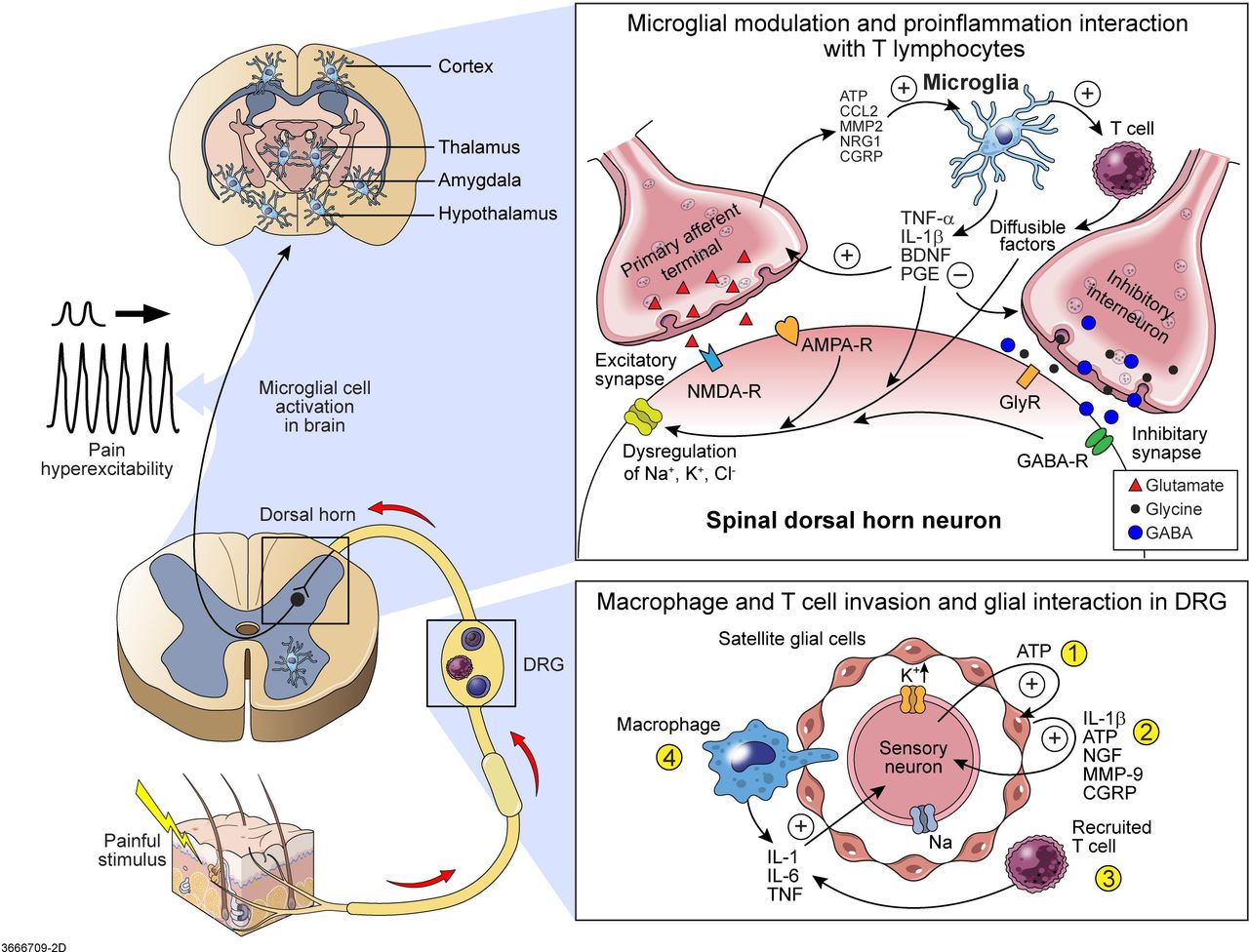
Glial and immune cell regulation of pain. Satellite glia and microglial cell activation lead to peripheral and central pain sensitisation.10 (Top right) The glial-mediated mechanisms that facilitate excitatory synapses include increased glutamate release, glutamate receptor activation and increased Na+ current. Pain from primary afferent terminals leads to release of ATP, CCL2, MMP2, NRG1 and CGRP, resulting in microglial activation. T cells are recruited and activated by microglia. BDNF is secreted by activated microglia, together with IL-1, ATP and MMP-9 secreted by satellite glial cells, bind to lamina I nociceptive neurons and trigger a shift of chloride anion gradients through potassium and sodium channels to produce hyperexcitability of the neuron central sensitisation. The suppressing mechanism in inhibitory synapses includes reduced release of GABA and glycine, inhibition of GABA receptors and reduced K+ current. (Bottom right) Dorsal root ganglia glial satellite cells activated by pain leads to peripheral sensitisation. ① Sensory neurons release ATP to activate satellite glial cells. ② Satellite glial cells then release IL-1β, ATP, NGF and MMP-9 to sensitise sensory neurons, eliciting spontaneous discharge of action potential. ③ Resident macrophages and T cells in the DRG release chemokine fractalkine and CCL2, triggering more macrophage and T-cell recruitment. Macrophages and T lymphocytes increase the synthesis and release of cytokines IL-1, IL-6 and TNF. TNF increases the density of tetrodotoxin-resistant Na+ channel currents in nociceptors (sensory neuron). ④ Macrophages pass though the satellite cell sheath around the primary sensory neuron in an attempt to clear damaged neurons. As a result, extracellular K+ levels of sensory neurons are dysregulated, leading to neuronal hyperexcitability. (Top left) Chronic pain can lead to brain microglia proliferation and activation at the cortex, thalamus, amygdala and hypothalamus, with resultant central pain sensitisation. AMPA-R, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ATP, adenosine triphosphate; BDNF, brain-derived neurotrophic factor; CCL, C-C motif ligand; CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglion; GABA-R, gamma-aminobutyric acid receptor; GlyR, glycine receptor; IL, interleukin; MMP-9, matrix metalloproteinase 9; NGF, nerve growth factor; NMDA-R, N-methyl-D-aspartate receptor; NRG1, growth factor neuregulin-1; TNF, tumour necrosis factor.
Microglial cells directly contribute to central sensitisation; however, glia within the peripheral nervous system (PNS) also have a role in the sensitisation of nociceptive signalling (figure 2).10 Dorsal root ganglia (DRG) glial satellite cells are activated by sensory neuron release of ATP. Satellite glial cells are thereby stimulated to release IL-1β, ATP, nerve growth factor, metalloproteases and CGRP, which sensitises sensory neurons eliciting spontaneous action potential discharges. Next, resident macrophages and T cells in the DRG releasing chemokines trigger more macrophage and T-cell recruitment. Macrophages and T lymphocytes increase synthesis and release of cytokine IL-1, IL-6 and tumour necrosis factor (TNF). TNF increases the density of tetrodotoxin-resistant Na+ channel currents in nociceptors (sensory neurons). Macrophages pass though the satellite cell sheath around the primary sensory neuron in attempt to clear damaged neurons. As a result, extracellular K+ levels of sensory neurons are dysregulated, leading to neuronal hyperexcitability.
Adaptive immunity and pain
Pain can also occur as a result of direct IgG-induced injury of nociceptive fibres via molecular mimicry, either post infectious (eg, pain with Guillaine-Barré syndrome from Campylobacter jejuni infection) or with sterile antigen exposures, which may break immunological tolerance (eg, aerosolised porcine neural tissue exposure and cancers in paraneoplastic syndromes, table 2).15 Integral in the generation of antigen-specific IgGs are the initial innate interactions of neutrophils, eosinophils, macrophages and T cells (figure 1). The specific neuronally derived signalling molecules help drive the adaptive T-cell response and propagation of specific IgG reactions. The process begins with the detection, engulfment and phagocytosis of a specific antigen by an antigen-presenting cell (macrophage, dendritic cell, B cell, others), with subsequent presentation to naive T and B cells. Naive T cells then recognise these processed antigens on MHC molecules and mature into different subtypes driving the immune response. Depending on the type of antigen and the specific cytokine reaction, naive T cells mature into different subtypes driving the immune response to propagation and/or tolerance of the targeted antigen.
Autoantibodies in chronic pain
Poor correlation of pain with extent of injury, as measured by location and extent of axonal loss, raises questions as to cause of many painful conditions. An emerging concept of pain relates to the observation that IgG complexes can directly bind Fc-gamma receptors on nociceptive fibres in the DRG or their soma without axonal loss.16 Specifically, immunoglobulins can stimulate nociceptive receptors by either binding the constant region (Fc) or antigen-binding (Fab) regions without inflammation at nodal or soma structures.
Autoimmune IgG-mediated neurological pain disorders
Recent work in the field of immunology has led to the recognition of a group of autoimmune pain disorders caused by specific IgGs directed against antigens in the somatosensory pathway. They are distinguished in part by their typical lack of neural tissue destruction as occurs in vasculitis, where no IgG autoantibody has been identified as causal. Among all chronic pain disorders, patients with ‘autoimmune pain’ are rare and typically have other neurological accompaniments beyond pain with some having cancer responsible via a paraneoplastic mechanism.15
A number of clinical features can point to considerations of ‘autoimmune pain’ disorders, including the temporal onset, neurological localisations and associated neurological and psychological accompaniments. If suspected, laboratory testing can confirm the diagnosis through identification of a specific causal autoantibody. Patients typically have subacute onsets with multifocal neurological manifestations including both PNS and CNS involvements. Neurological involvements include (1) small fibre sensory (heat, pain, cold) either loss and/or exaggerated function, (2) autonomic dysfunction (eg, orthostatic/cardiogenic and/or gastrointestinal dysmotility with sudomotor sweat loss or hyperhidrotic attacks) and (3) encephalopathy with or without seizures or paroxysmal spells. Frequently, MRI or EEG equivalents are not found until late in the disease. Similarly, routine nerve conductions are usually normal, but skin surface electromyography (EMG) can demonstrate cramp discharges or hyperexcitable discharges on needle EMG. Autonomic testing (gastrointestinal motility–gastric emptying, sudomotor sweat tests and cardiovagal changes) involvements may also be present. Sural nerve biopsies are generally not helpful, apart from excluding other pain mechanisms related to destructive injury of peripheral nerves, that is, vasculitis and amyloidosis. Epidermal skin biopsies immunostained with antibodies against PGP9.5 that resides within small unmyelinated fibres can help quantify nerve fibre involvements.
IgG-mediated pain disorders have overlapping pain symptoms, including cramps, spasms, burning, paresthesia, lancinating pain and itch. Below are examples having divergent targeted antigen-specific forms. We do not include painful disorders where specific IgGs and mechanistic antigens in pain are not yet known, such as vasculitis, contact and atopic dermatitis, herpes zoster nor disorders where axonal loss from direct immunoglobulin destruction of nociceptive fibres occurs like in CRMP5 painful neuropathy.17
Pain in VGKC complex (LGI1-IgG, CASPR2-IgG) and CASPR1-IgG autoimmunity
The voltage-gated potassium complex (VGKC)-IgG disorders provided an initial example of ‘autoimmune pain’.18 19 VGKC-IgG autoimmunity was first described with painful muscle cramps, spasms and fasciculations (spontaneous contraction of a small number of muscle fibres, causing a flicker of movement under the skin) with electrographic EMG hyperexcitability with myokymia (repetitive regular firing bursts of motor units, 20–80 Hz), cramp discharges and neuromyotonia (rapid firing long continuous firing motor units, 150–300 Hz) in Isaacs’ syndrome (acquired neuromyotonia with hyperhidrosis) and Morvan syndrome (neuromyotonia, dysautonomia, limbic encephalitis and sleep disturbance). Subsequently, it was realised that leucine-rich glioma-inactivated 1 protein (LGI1) and contactin-associated protein-2 (CASPR2) are the primary antigen targets within the VGKC-IgG complex (figure 1).20 21 Other neurological hyperexcitability features, apart from those involving pain somatosensory pathways, most commonly are limbic encephalitis, seizures, faciobrachial dystonic seizures (LGI1-IgG specific), paroxysmal dizzy spells and dysautonomia including gastric dysmotility.20–22 Cancer is actually uncommon, but when found, it is most likely linked to thymoma, with CASPR2 autoimmunity.22 23
In patients with LGI1 and CASPR2 seropositivity, pain is often described beyond those areas of motor hyperexcitability recordable by EMG. Hence, small nociceptive fibre hyperexcitability and dysfunction were theorised and objectified by thermoregulatory sweat testing, quantitative sudomotor reflex screens (showing hyperhidrosis) and quantitative sensory testing (showing heat pain hyperalgesia).22 Patients typically describe burning and stinging pain in the extremities, less commonly the trunk and face. Pathological skin biopsies, epidermal laser-evoked potentials and whole sural nerve biopsies from CASPR2-IgG and LGI1-IgG seropositive patients demonstrate no (or limited) interstitial abnormalities and often normal fibre counts, supporting primary nociceptive hyperexcitability as the cause of pain.24 25 Among 256 patients who were LGI1-IgG or CASPR2-IgG seropositive (or both), neuropathic pain was documented in 21% with LGI1-IgGs and 46% with CASPR2-IgGs.22 Of patients undergoing EMGs, 24% of the LGI1-IgG–positive patients and 75% of CASPR2-IgG–positive patients had electromyographic cramps (involuntary motor unit firing in a large area of muscle up to 150 Hz, typically painful), fasciculations, myokymia (continuous bursts of 2–10 motor unit potentials firing at 40–60 Hz) or neuromyotonia (motor unit potentials firing at 100–300 Hz, often with varying amplitude). Pain was often severe, and opioid use was common (38% of LGI1-IgG and 86% of CASPR2-IgG–positive pain patients). Pain rehabilitation referral was requested for 25% of patients. In this series and earlier series, the immune responsiveness of pain and the other neurological features is often dramatic; however, many need long-term immunotherapy and/or antiepileptic medications.19 23 24 26 Varied immunotherapies have been used successfully including steroids, intravenous immunoglobulin (IVIG), methotrexate and others in open trials.
The understanding for molecular mechanism of how antibodies in the VGKC complex cause pain is increasing.27 CASPR2 belongs to the neurexin family, localises to the juxta-paranode and interacts with contactin-2 of myelinated axons in both PNS and CNS. It is not restricted to the axons, also present at the soma membrane of DRG cells. Earlier work with sera from VGKC-IgG–seropositive, patients with Isaacs’ syndrome (presumed CASPR2-IgG) showed neuronal culture suppression of voltage-gated outward K(+) current, with hyperexcitability associated with an observed decrease in VGKC channel density at the node.28 Passive transfer of CASPR2 antibodies from patients into mice demonstrates direct antibody binding to the afferent cell bodies in the DRG with resultant decreased expression and function of Kv1 channels at the soma membrane and reduced nodal localisation. These results link CASPR2 antibodies to enhanced nociceptor hyperexcitability and pain.29 Additionally, CASPR2 antibodies have also been implicated in causing central microglia reactivity following exposure in utero (but not in adulthood when the blood–brain barrier is intact).30 These findings may have neurodevelopmental implications in terms of central sensitisation abnormalities mediated by excess microglial activation. LGI1 prevents rapid Kv1-channel inactivation within the neuronal presynaptic membrane and is highly expressed within the brain and spinal cord. The locus of action of antibodies directed against LGI1 is not yet known, but pain could be mediated by potentiation of synaptic transmission within somatosensory pathways.
Contactin-associated protein 1 (CASPR-1) exists separately from the VGKC complex at the paranodal region (figure 1). It has recently been described as a rare novel antigen with autoimmunity and neuropathic pain in association with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and Gullain-Barré syndrome.31 Sensory deficits and severe pain in the back and feet can develop at the onset in CASPR1-IgG–positive patients. Serum from patients with CASPR-1 autoantibodies bound to small-diameter fibres without reduction of intraepidermal nerve fibre density, suggesting a functional mechanism for the pain. Complement binding was not evident, consistent with the characteristics of IgG4 subclass. This group of patients responded poorly to IVIG, but resolved completely after treatment with rituximab (B-cell depletion treatment).32
The protein complex formed by CASPR1, contactin-1 (both axonal) and neurofascin 155 (NF155; Schwann cell) organises as septate-like tight junctions at the paranodal loops of the node of Ranvier and is essential to maintain molecular organisation of the node of Ranvier in all myelinated fibres (figure 1). Interestingly, pathogenic antibodies against contactin-1 and NF155 occur in a subset of patients with CIDP without neuropathic pain.33 34 Because CASPR1 regulates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors centrally,35 central sensitisation could play a role in this selective pain component with CASPR1 autoimmunity, but this has not directly been studied.
Pain and neuromuscular hyperexcitability in other ion channels (N and P/Q VGCC-IgG, nicotinic and muscarinic AChR-IgG)
P/Q-type and N-type voltage-gated calcium channels (VGCCs) are well-known targets in Lambert-Eaton syndrome. They are also associated with other neurological phenotypes, including pain with stiff person syndrome (SPS) and neuropathy with tumours besides small cell lung cancer, which is most common.36–38 Neural tissue loss is not apparent (or minimal) relative to pain severity. In a review of different ion-channel antibodies in 77 patients with neuromuscular hyperexcitability (35 neuromyotonia, 32 cramp-fasciculation syndrome, 5 rippling muscle syndrome and 5 focal neuromuscular hyperexcitability) versus 85 controls, the P/Q and N VGCCs were found in 12% versus 0% controls. Also seen was 35% with VGKC, 12% with ganglionic nicotinic acetylcholine receptor (AChR) antibodies (alpha-3) and 16% muscle AChR antibodies. The majority had painful neuromyotonia. The calcium channels are expressed by DRG cells and localise presynaptically within laminae 1 and 2 of the dorsal horn of the mouse spinal cord, where they modulate the influx of calcium during the release of neurotransmitters such as glutamate and substance P, and these channels have an important role in promoting the excitability of nociceptive neurons.39 Therefore, although VGCCs, ganglionic nicotinic AChR and muscle AChR are an unproven cause of pain, there are theoretical reasons to continue considering their potential role in autoimmune pain syndromes.
IgG targets in painful cramps and spasms
Painful cramps and spasms can occur with immune-mediated peripheral or central hyperexcitability. These are paroxysms, in specific muscle groups, for example, symmetric ‘plantar flexor spasms’ most commonly central in origin, or in isolated asymmetric muscles such as calves, finger flexors and oris oculi, which are more commonly peripheral in origin. Cramp discharges (either peripheral or central) are resultant from neural or muscle fibres firing simultaneously.
Stiff person syndrome and progressive encephalitis with rigidity and myoclonus (GAD65, GlyR, amphiphysin-IgGs)
Antibodies to glutamic acid decarboxylase (GAD) are associated with forms of stiff persons syndrome spectrum disorders (SPSDs).40 41 SPS is a rare, acquired, autoimmune neurological entity characterised by progressive fluctuating muscle stiffness (rigidity): 59.4% with cramps or spasms and 33.3% with pain.42 Pain, especially in the lower back or legs, may be the initial experience. Painful spasms may occur spontaneously or be triggered by a sudden noise or light physical contact. GAD65 IgG values >200 nmol/L are common and in one large series the median value was 623 nmol/L.43 For all neurological presentations of GAD65, only ~50% have a good response to immune treatment.44 Despite many patients not having a response to immune treatment, the γ-aminobutyric acid agent like diazepam can still be symptomatically beneficial, but often at high dose, that is, 40.0 mg/day.43
GAD has two isoforms, GAD65 and GAD67, and both are CNS gamma-aminobutyric acid (GABA) synthetic enzymes that convert l-glutamate to GABA. GAD65, which has greater autoantigenicity, is expressed in presynaptic GABA-ergic neurons for vesicle release, whereas GAD67 preferentially synthesises cytoplasmic GABA. Pain in SPSD is unlikely caused by peripheral neuropathy, as classic GAD65 antibody-positive peripheral neuropathy is not well substantiated, mainly large fibre neuropathy and commonly coexisting with diabetes mellitus.45 46 GAD65 antibody inhibits GAD65 activity and disrupts GABA synthesis in vitro.47 In rat and mouse models of chronic pain, GAD65 transcription is downregulated, thereby impairing GABA synaptic inhibition in the brain stem nucleus raphe magnus and promoting the excitability of pain-facilitating neurons.48 Thus, hyperactivated neurons in the CNS nociceptive pathway likely are the direct cause of SPS and pain. Antibodies to GAD65 occur in 60% to 70% of SPS cases, but SPS is also associated with antibodies to GlyR (15.9%),49 amphiphysin (<5% cases, in the setting of both small cell lung and breast cancers)49 and gephyrin (one case).50
Recently, GlyR autoantibodies have also been detected in serum and cerebrospinal fluid of patients with progressive encephalitis with rigidity and myoclonus (PERM). PERM has characteristics of acute or subacute painful spasms and muscle stiffness, and is considered when painful spasms are rapidly progressive, widespread and also involve the brain stem and spinal cord. Painful spasms and rigidity of the neck, back and legs are the most common feature of PERM.51 GlyR autoantibody may coexist with other antibodies, such as those against N-methyl-D-aspartate receptor, GAD-65, VGKC complex, myelin oligodendrocyte glycoprotein and aquaporin-4. GlyRs are pentameric ligand-gated chloride channels widely expressed in CNS, but transcripts of GLRA3 (encoding GlyR subunit α3) are found only in the superficial layers of the spinal dorsal horn and in the cerebellum and olfactory bulb. The autoantibodies disrupt α3-containing GlyRs in the dorsal horn, resulting in reduced glycinergic neurotransmission. Therefore, disinhibition may explain the increased pain and itch perception in patients with PERM who are GlyR-IgG positive.51 52
Another presynaptic antigen pertinent to SPS-like disorder is amphiphysin. Amphiphysin is critical for recycling synaptic vesicles after GABA release. Amphiphysin-IgG autoantibodies are rare, but highly associated with paraneoplastic neurological disorders.53 Anti-amphiphysin antibodies might induce SPSD by inhibiting the function of GABAergic neurons.54 The potential immune effector is local reactivation of amphiphysin peptide-specific cytotoxic CD8+ T cells throughout the brainstem, spinal cord parenchyma and DRG. This may explain the very poor immunotherapy response in these patients.
Neuromyelitis optica and aquaporin-4-IgGs
Pain is often a major symptom associated with neuromyelitis optica (NMO), and pain remains intense, even with immunotherapy and lesional burden management, sometimes with nominal neurological deficits.55 56 The pain is disproportionately high in patients with NMO when compared with patients with multiple sclerosis with similar lesion burden.
Two pain types are characteristic in NMO: (1) painful tonic spasms, especially in the recovery stage of NMO, affecting up to one-third of patients with anti-AQP4 autoantibodies; and (2) extremity neuropathic pain.57 Neuropathic pain is particularly associated with persistent thoracic spinal lesions.58 The AQP4 water channel IgG target is believed to account for the disproportionate extent of pain.59 Astrocyte injury in NMO is mediated by AQP4 IgG1 antibodies-activated complement or the interaction of effector cells and NFⱪB driving progranulocyte astroglial response, that is, in part an indirect inflammatory cause of pain.60 61 The AQP4 channel is distributed widely in descending and ascending nociceptive somatosensory pain modulating structures (figure 1).62 Autoantibodies against AQP4 are believed to disturb the periaqueductal grey and rostroventral medulla pathways. Glutamate is the principal excitatory neurotransmitter in the first synaptic relay of nociceptive pathways in laminae I and II of the spinal dorsal horn. In established NMO lesions, loss of astrocytes inevitably interrupts the glutamine–glutamate–GABA pathway, which is important in regenerating levels of GABA. Furthermore, downregulation of AQP4 and its complexed EATT2 transporting after binding of AQP4-IgG results in an increase of extracellular glutamate. Such excess glutamate accumulation in the extracellular space would induce aberrant neuronal excitation, resulting in pain. Activation of the complement system with resulting production of both C5a and C5b will injure the astrocyte through membrane attack complex formation and create an inflammatory microenvironment, promoting pain (figure 3).63

Pain excitation mechanisms in AQP4 autoimmunity. Pathogenesis of AQP4-IgG-mediated neuromyelitis optica lesions and impactors of pain development. (Right) IgG-AQP4-EAAT2 modulation with internalisation stimulates extracellular glutamate production, leading to long-lasting pain amplification. Glutamate is the principal excitatory neurotransmitter in the first synaptic relay of nociceptive pathways in laminae I and II of the spinal dorsal horn. Segmental and descending inhibition is also impaired. (Left) Antibody-dependent cellular cytotoxicity leads to astrocyte injury and loss of inhibitory synaptic input. Loss of astrocytes, which are an exclusive source of glutamine in the central nervous system, interrupts the glutamine–glutamate–GABA axis, with neural hyperexcitability theorised. (Bottom) Complement activation leads to excitotoxicity and neural activation by cell-mediated cytokine chemokine inflammatory processes. The combination of events leads to interruption of the glutamine–glutamate–GABA pathway, which is essential in pain modulation. AQP4, aquaporin-4; ATP, adenosine triphosphate; CCL, chemokine (C-L motif); CXCL, chemokine (C-X-C motif); EAAT2, excitatory amino acid transporter 2; GABA, gamma-aminobutyric acid; IL-17, interleukin-17; P2×3, purine receptor 2×3; TNF, tumour necrosis factor.
Reducing further loss of astrocytes by prophylactic immunotherapy is foremost in NMO. Anecdotal evidence has also suggested that carbamazepine is frequently helpful, but how the drug’s mechanism of action (prevention of inward flux of neuronal sodium) diminishes pain is unclear.64
Autoantibodies associated with complex regional pain syndrome
Complex regional pain syndrome (CRPS) is characterised by limb-confined, spontaneous and/or stimulus-induced pain (burning pain with allodynia and hyperalgesia), which is usually out of proportion to a precipitating minor injury, accompanied by autonomic dysfunction (changes in vascular tone, sudomotor function, skin temperature and oedema), motor disturbances (weakness, tremor and muscle spasms) and trophic skin changes with bone demineralisation.65 Biopsies have shown minimal cellular infiltration, although a network of pro-inflammatory mediators (TNF-α, IL-1β, IL-6, IL-8 and nerve growth factor (NGF)) may be contributory through nociceptor sensitisation.66 Circulating IgG autoantibodies against autonomic nervous system structures have suggested that CRPS may be an IgG-mediated pain disorder.67 This experience and pathological observations have led others to propose an alternative term for CRPS, that is, ‘IRAM’ (injury-triggered, regionally restricted autoantibody-mediated autoimmune disorder with minimally destructive course).68
In CRPS, the profound, sympathetically maintained pain is considered a defect in the sympathetic–sensory decoupling mechanism.69 IgG autoantibodies with agonistic-like properties on the β2-adrenergic receptor (β2AR) and/or the muscarinic-2 receptor (M2R) or activating antibodies to alpha-1a adrenoreceptors were found in patients with CRPS.68 70 Both β2AR and M2R are involved in modulation of pain and in inflammation.71 Under physiological conditions, activation of M2R on peripheral nociceptors inhibits nerve excitability.72 M2R autoantibodies are capable of promoting pain through nociceptive hyperexcitability in CRPS.73 The discovery of these autoantibodies indicated that patients might respond to immunotherapy, but a large (N=108 patients) multicentre randomised blinded placebo-controlled trial with 0.5g/kg IVIG over 6 weeks could not confirm benefit compared with placebo-treated patients.74 It is likely the discovered autoantibodies are not the cause of the syndrome; rather, they may be biomarkers of a complex and poorly understood pathogenic process.
Biological and emerging designer immune therapies for pain
New designer immune-based biologic therapies are emerging from the increasing understanding of the molecular pathways associated with pain. Antibody therapies are most attractive for pharmaceutical development when a protein exerts a strong negative affect on a studied function such as pain excitation. Antibody therapies have high specificity with reduced off-target effects, which is not possible in many non-biologic therapies. The areas where antibody designer therapies are furthest along in pain evaluation are in two well-studied inflammatory molecular innate pathways: (1) calcitonin gene-related peptide in headache and (2) NGF in a host of disorders including arthritic pain (figures 2 and 3).
Anti-inflammatory biological therapies can repress the innate-immune mechanisms of pain. However, most available biologicals targeting the inflammatory cascade have yet to be evaluated in clinical trials for pain relief, including antibodies targeting B cells (anti-CD20 and CD19 antibodies) or cytokines (IL-1-anakinra, IL6-toclizumab, TNF-α-infliximab, etanercept and adalimumab), complement inhibition (C1-esterase inhibitors, anti-C5 mAbs) and proteasome inhibitors (bortezomib, carfizomib, ixazomib). With one exception, complement interference by soluble human complement receptor type-1 (TP10) has been studied in phase II clinical trials to treat chest pain complications of cardiopulmonary bypass surgery related to complement activation.75
Anti-NGF therapy
NGF is a potent neurotrophic factor during mammalian embryogenesis produced in limited amounts by innervation targets and required for the survival and development of nociceptors and sympathetic efferents. In the postnatal period and adulthood, NGF has been found to have important sensitising effects on nociceptors and administration promotes mechanical and thermal hyperalgesia in rodents and indeed humans.76 This is important because inflammation either of injured tissue or nerve results in increased expression of NGF, and this factor makes a major contribution to inflammatory pain.77 Its high affinity receptor, tropomyosin receptor kinase A (trkA), is expressed by peptidergic nociceptive afferents and mast cells. Following binding of NGF to trkA, the receptor autophosphorylates and activates multiple downstream signalling pathways in nociceptor terminals, activating key transducers such as TRPV1, a ligand-gated ion channel that responds to noxious heat and capsaicin. Furthermore, the receptor complex is internalised at nerve terminals and transported to the DRG where it affects transcription regulation of numerous cell surface receptors and ion channels, important in pain regulation, as well as neuronal interactions. NGF binds to trkA receptors on mast cells and can drive mast cell degranulation and release of inflammatory mediators. The downstream effects of enhanced NGF signalling are both peripheral and central sensitisation, resulting in chronic pain (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Nerve growth factor nociceptive pain regulation. NGF is a potent regulator of nociceptive pain thresholds. NGF is released by diverse tissue injuries, including those involving viscera, bones and nerve.77 Recent work suggests that it is a potential target for biological antibody therapy in the trigeminal sensory nerve pathway of headache but also other pain generators (top). NGF can cause heightened pain threshold by directly binding to mast cells, causing degranulation and increased NGF production. Local modulation alters ionic transport regulators of pain, leading to membrane excitability (bottom). Once bound to its receptor (tropomyosin-related kinase A) at sensory nerve terminals, NGF undergoes retrograde transport and cause increased transcription of a number of inflammatory neuropeptides. Some of these neuropeptides undergo anterograde transport back to the distal sensory terminals (A, central sensitisation) or rostrally to the spinal cord and brain (B, peripheral sensitisation). BDNF, brain-derived neurotrophic factor; BR2, bradykinin receptor 2; CGRP, calcitonin gene-relatedpeptide; DRG, dorsal root ganglia; NGF, nerve growth factor; P75, neurotrophin peptide 75; SP, substance P; TrKA, tropomyosin kinase-A; TRPV1, transient receptor protein vanilloid-1.
Immunotherapy trials have focused on targeting circulating NGF with NGF-sequestering agents to prevent NGF from binding to trkA, or to inhibit trkA function.78 Preclinical, phase I, II and III clinical trials have been completed for anti-NGF antibodies.79 A phase III study of 7000 patients with osteoarthritis, chronic low back pain or cancer pain is underway for the drug tanezumab with fast-track designation by the US Food and Drug Administration (www.clinicaltrials.gov). Multiple earlier trials showed encouraging results, with superior performance of this drug and two other anti-NGF drugs (fulranumab and fasinumab) over placebo. One large double-blinded, placebo-controlled study (N=610) showed that tanezumab was superior to non-steroidal anti-inflammatory drugs and opioids in terms of functional outcomes and pain scale improvements.80 An unresolved issue is the potential for accelerated osteoarthritis, which appears to progress more rapidly in persons on drug compared with controls. Safety studies did not demonstrate aberrant effect on peripheral nerves. Limited neuropathic pain investigation trials have also been completed with diabetic neuropathy, visceral pain (pelvic floor and prostatis) and post herpetic neuralgia pain, and all have shown promise.78 79
Anti-CGRP) therapy
CGRP, part of the calcitonin family of peptide hormones, has two isoforms (α and β). The α isoform is a 37-amino-acid peptide that is released in the trigeminal ganglion after migraine triggers (eg, toxic, ischaemic, metabolic and inflammatory insults). Once released, it is a potent vasodilator in the meninges and is associated with release of nitrous oxide. CGRP can promote nociceptive signalling by a number of mechanisms, including (1) stimulation of satellite glial cells (figure 2), (2) excitation of second-order neurons in the trigeminal nucleus (central sensitisation) and (3) excitation of primary afferent terminals (peripheral sensitisation).81 The subunit is expressed in keratinocytes and enteric neurons of the gut and is a potential drug target for other pain disorders.
Evidence is growing from migraine clinical trials that monoclonal antibodies to alpha-CGRP may benefit migrainors.82 There are currently four CGRP monoclonal antibodies under phase III trials (eptinezumab, galcanezumab, frestanezumab, erenumab), all but erenumab target the CGRP directly. The preceding safety trials showed a modest reduction in the number of days of headache and in daily analgesic intake, supporting an important role for CGRP in the pathogenesis of migraine pain. These early studies showed a safety profile that was similar to a placebo, but the phase III trials will more clearly assess safety and efficacy.
Practical aspects of pain management in affected patients
For autoimmune pain disorders, as more mechanistic drugs become available, greater choices will be afforded to physicians. However, we will always have to balance our desire to help patients with the limitations and risks of therapies. This is especially emphasised because the inherent risks of immunotherapy are great. The first step is to ensure the accuracy of a diagnosis that is immune responsive. Internationally, it is now possible to order accurate testing from certified laboratories for most of the antibodies discussed. Next, it is important to create a standard outcome measure for immunotherapy. It can be difficult in pain, as objective outcome measures are especially difficult for this subjective experience. The visual analogue pain scale is validated useful and a sustained 50% improvement is an excellent indication for a meaningful improvement. For patients on high-dose steroids, euphoric effect from the drug can impair pain assessment. Nevertheless, starting with high-dose steroids then moving to lower doses and eventual steroid-sparing agents is a common approach, especially when other devastating neurological features are present. In patients with LGI1 or CASPR2 autoimmunity, the resultant seizures, encephalopathy, memory difficulties and pain often improve simultaneously during immunotherapy.19 In contrast, for patients with pain and NMO, immunotherapy for maintaining spinal cord and eye health may not immediately lead to improved pain, and the goal of immunotherapy may be to avoid future spinal cord and optic nerve injury.
It is helpful to realise that an earlier injury in the somatosensory pathway without active autoimmunity may drive ongoing pain, which is more common in AQP4, GAD and amphiphysin than LGI1 and CASPR2 autoimmunity. In all patients, membrane-stabilising drugs can be very helpful as these drugs have nociceptive membrane-stabilising features.26 When selecting a specific type of drug, cost, tolerance and efficacy all are important factors. First-line agents for neuropathic pain appropriate to these disorders include gabapentinoids, tricyclic antidepressants and selective norepinephrine, serotonin reuptake inhibitors.83 Physicians should allow patients to control the rate of escalation to avoid intolerance from commonly somnolence and altered sensorium, within maximal dosing guidelines, before moving to other medications. For patients with NMO with cramps and spasms, carbamazepine can provide selective advantage if gabapentin is ineffective. Narcotics should only be prescribed sparingly, barring rescue scenarios, given the chronic nature of these disorders and lack of evidence on functional improvements of narcotics for neuropathic pain.4 Behavioural and psychotherapeutic approaches including coping strategies should all be attempted and common coexisting depression addressed.84 Trial and error for various treatment options while constantly weighing risks and benefits, and engaging patients in the decisions are generally appropriate and beneficial.
Conclusions
Autoimmune pain disorders, in which specific immunoglobulin antigen targets are within the nociceptive pathways, are rare but important to recognise because they are often immune therapy responsive and are typically associated with other neurological deficits and possibly cancer. Improved understanding of the mechanisms of pain generation is facilitating developments of designer biological medications for pain management. Membrane-stabilising epilepsy drugs can help patients with relatively limited risk, whether pain is associated with non-specific inflammation or directed immunoglobulins against different portions of the somatosensory nociceptive pathway. The discovery of specific circulating autoantibodies needs to be critically assessed before immune causality for pain is assumed. International collaborative efforts are needed, with emphasis on conducting multicentre blinded trials and assessing the most beneficial disease management options for patients with these rare disorders.
References
Footnotes
Correction notice Since this article was first published online the affiliations for Dr Watson and Dr Klein have been updated.
Contributors XM and CJK created the draft, selected reports and extracted the data. DLHB, LAQ, L-JW, SRI, JCW and SJP all critically contributed in planning and editing the manuscript and approved the final version. CJK is the guarantor of the manuscript.
Funding Mayo Foundation supported Mr Steven D Orwoll who is the medical illustrator who assisted in producing the figures.
Competing interests DLHB reports that he has acted as a consultant on behalf of Oxford Innovation for Abide, Biogen, GSK, Lilly, Mitsubishi Tanabe, Mundipharma and TEVA over the last 3 years. LAQ reports that he has provided expert testimony for Grifols and CSL Behring and received research funds from Novartis Spain and Grifols. SRI reports that he is a coapplicant and receives royalties on patent application WO/2010/046716 (UK patent no. PCT/GB2009/051441) entitled ‘Neurological Autoimmune Disorders’. The patent has been licensed to Euroimmun AG for the development of assays for LGI1 and other VGKC-complex antibodies. The remaining authors report no relevant disclosures to this work.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.