Article Text

Abstract
Advances in neuroimaging are ideally placed to facilitate the translation from progress made in cellular genetics and molecular biology of neurodegeneration into improved diagnosis, prevention and treatment of dementia. New positron emission tomography (PET) ligands allow one to quantify neuropathology, inflammation and metabolism in vivo safely and reliably, to examine mechanisms of human disease and support clinical trials. Developments in MRI-based imaging and neurophysiology provide complementary quantitative assays of brain function and connectivity, for the direct testing of hypotheses of human pathophysiology. Advances in MRI are also improving the quantitative imaging of vascular risk and comorbidities. In combination with large datasets, open data and artificial intelligence analysis methods, new informatics-based approaches are set to enable accurate single-subject inferences for diagnosis, prediction and treatment that have the potential to deliver precision medicine for dementia. Here, we show, through the use of critically appraised worked examples, how neuroimaging can bridge the gaps between molecular biology, neural circuits and the dynamics of the core systems that underpin complex behaviours. We look beyond traditional structural imaging used routinely in clinical care, to include ultrahigh field MRI (7T MRI), magnetoencephalography and PET with novel ligands. We illustrate their potential as safe, robust and sufficiently scalable to be viable for experimental medicine studies and clinical trials. They are especially informative when combined in multimodal studies, with model-based analyses to test precisely defined hypotheses.
- dementia
- MRI
- PET
- image analysis
- functional imaging
Statistics from Altmetric.com
Introduction
Brain imaging can bridge the gap between the progress made in understanding the cellular genetics and molecular biology of neurodegeneration1 w61–63 and clinical trials of novel interventions for dementia (box 1). The success of such translational medicine will be measured in terms of better diagnosis, treatment and ultimately prevention.
Highlights
Neuroimaging can be used to establish and test models of disease mechanisms in humans.
Positron emission tomography can quantify and localise molecular processes in vivo. Amyloid imaging has already changed clinical trials design and identified new drug targets.
New ligands for synaptic density, protein synthesis, tau and other proteins are scientifically informative but have yet to find their place in healthcare.
Neuronal populations are functionally and structurally connected at multiple scales, which can be examined by multimodal brain imaging.
Relating molecular pathology to brain connectivity reveals disease mechanisms and validates drug targets.
Focal neurodegenerative syndromes are important disease models, selectively perturbing complex neuronal systems.
Powerful model-based analyses can reveal microcircuit-level consequences of neurodegeneration, in humans.
Neuroimaging can enrich and stratify cohorts, for precision medicine approaches.
Small-N experimental medicine studies and large-N observational trials enable the generation and testing of complementary hypotheses.
Data sharing is now readily available, facilitated by consensus data formats and infrastructure like the Dementias Platform UK Portal, enhancing the value of imaging data through Open Data initiatives, meta-analysis and repurposing. Disease-specific examples such as Alzheimer’s Disease Neuroimaging Initiative and Parkinson Progression Marker Initiative, as well as global initiatives such as ENIGMA have transformed the field of collaborative research.
Clinical trials can build on the success of longitudinal cohort studies combining behavioural and cognitive assessment with multimodal imaging, genetics, serum and cerebrospinal fluid measures.
For diagnosis, improving current clinical practice requires quantitative methods that are accurate in terms of individual disease processes, and allow precision medicine by accommodating the complex multidimensionality of dementia. This multidimensionality is recognised in psychiatry in the Research Domain Criteria Criteria,2 providing a conceptual framework to integrate pathophysiology and symptomatology in spectral features of disease, rather than arbitrary categories. The spectral nature of dementia phenotypes is increasingly recognised in trans-diagnostic cohort studies3 w64 65 and recent diagnostic criteria that encompass phenotypic variants.4 w66–71
For treatment trials, there are complementary roles of imaging. Imaging can identify individuals who are well but at-risk for dementia, for genetic or unknown reasons, tracking latent or premanifest pathology.5–7 w72–74 Imaging also supports experimental medicine studies, with surrogate markers of disease processes as treatment outcomes. These secondary outcomes include evidence of drug target engagement, with diverse measures of neuropathology, neurophysiology, connectivity and function.
For prevention, imaging allows insight into modifiable disease processes including neurochemistry, physiology, molecular pathology and structure, and how these interact with the environment and genetics. While no single imaging modality quantifes the whole cascade of events from root cause to final phenotype, combinations of imaging methods can connect each of these causal processes.8 9
The role of imaging to inform causal models of disease allows the design of rational, precise and optimally powered clinical trials. This can de-risk trials, with better designs and endpoints, including early closure of futile lines of enquiry so as to direct resources more effectively.
This review illustrates the advances in neuroimaging, beyond the structural imaging used clinically every day for diagnosisw75–78 and staging.10 11 w79–84 Novel ligands for positron emission tomography (PET) localise and relate molecular processes to each other in vivo. Combining this with connectivity analyses allows the direct evaluation of hypotheses of disease progression. The use of focal neurodegenerative syndromes as disease models allows the delineation of core neuronal systems, which can then be related back to help us understand the complex behavioural abnormalities that arise in dementia. Model-based analyses reveal microcircuit-level consequences of neurodegeneration in experimental medicine studies targeting specific disease mechanisms. With the advent of new therapies, neuroimaging is of greater utility than ever.
Quantifying molecular pathologies
Current ligands for PET allow the topographical quantification of metabolic activity ([18F]-fluorodeoxyglucose, FDG), beta amyloid (eg, PiB), tau neuropathology (eg, flortaucipir), neuroinflammation (eg, translocator protein (TSPO) and P2X7R ligands) and synaptic loss (eg, UCB-J).
In clinical practice, FDG reveals changes in regional metabolic activity that differentiate major dementia syndromes.w85 86 FDG-PET has largely superseded single photon emission computed tomography (SPECT) quantification of regional blood flow, due to its greater resolution, signal to noise ratio and robustness to non-linear relationships between metabolic demand and blood flow in cerebrovascular disease.w87–89 It can also be used as a trial outcome: tracking cerebral metabolic rate.w90
The synthesis of amyloid ligands (Pittsburgh compound B PiB, florbetapir, florbetaben and flutemetamol) enabled PET quantification of brain beta-amyloid.12 In clinical practice it is useful to segregate mild cognitive impairment (MCI) with underlying Alzheimer’s pathology from other causes.w91 92 It can also enrich study populations in clinical trials, screening out patients who are amyloid negative.w93 94 Amyloid PET has been used as gold standard for validation of biomarkers of Alzheimer’s disease (AD) in cerebrospinal fluid (CSF),w95 and blood.w96 97 However, amyloid burden stabilises by the time of diagnosis, and these ligands have little utility in tracking AD progression.w98–101
Longitudinal tracking of disease is more promising from ligands binding Tau.w102 103 Tau-aggregates are a defining feature of AD and FTLD-tau. In AD, the distribution of tau but not beta-amyloid determines phenotype13 and progression.w104 In molecular terms, beta-amyloid promotes the development and propagation of paired helical filaments of tau, which are especially neurotoxic in their oligomeric form.w105–107 This toxicity is amplified by the presence of beta-amyloid.w108 Several radioligands have been developed to assess regional Tau burden, including PBB3w109 and the THK-series,w110 but the most extensively evaluated is AV-1451, also known as T807 or flortaucipir.w111 112 This ligand has desirable properties in vitro, colocalising in postmortem samples with tau but not beta-amyloid, TDP-43 or α-synuclein.w113 It recapitulates Braak stages of AD,w114–116 and mirrors the regional distribution in focal subtypes.w117 AV-1451 binding is more closely linked to hypometabolism, atrophy and cognitive impairment than amyloid-PET.
Outside of AD, tau pathology characterises progressive supranuclear palsy (PSP),w118 corticobasal degenerationw119 and half of frontotemporal dementia (FTD).w120 121 Tau is also a modifier of Lewy body disease.w122 123 However, in non-AD diseases, the tau isoforms differ, lacking the characteristic ultrastructure of paired helical filaments in AD,w124–126 reducing binding affinity.w127 Despite relative in vitro insensitivity to these alternate isoforms, AV-1451 can identify the distribution of disease in PSP,14 CBSw128 and FTD due to mutations of the MAPT gene.w129 130
However, AV-1451 also has off-target binding, other than tau. For example, it recapitulates the distribution of pathology in semantic dementia,15 16 which is characterised by aggregation of TDP-43 in the absence of tau.w131 132 This lack of specificity is a barrier to stratifying cases of FTD according to molecular aetiology.w102 There remains an unmet need for specific PET ligands for FTLD-tau, TDP-43 and α-synuclein.
Neuroinflammation occurs in many neurodegenerative diseases, with geneticw133–135 and epidemiologicalw136 137 associations; and postmortemw138 139 and CSFw140 141 concomitance; matched in animal models.w142–144 This underlies an array of PET radiotracers for the neuroinflammatory cascade.17 The most established target is the 18 kDa TSPO, which is upregulated in activated microglia (and to some extent astrocytes). Increased microglial activation is seen in vivo in many neurodegenerative diseases,w145–150 and neuroinflammatory burden correlates with cognition.w151–153 Neuroinflammation is an early process in pathogenesis, preceding symptom onset in genetic cases,w154 and persisting through the disease course.w146–148 Second-generation TSPO ligands have improved signal-to-noise, but interindividual comparisons are confounded by genetic polymorphism.w155 Novel inflammation-related targets include the P2X7-receptor,w156 expressed by microglia and a therapeutic target in early AD.w157
PET ligands for new pathogenic mechanisms are emerging. Changes in synaptic density may precede atrophy and symptom onset,18 19 w158 and can now be quantified by targeting synaptic vesicle glycoprotein 2A, with ligands such as 11C-UCB-J. This reveals 20%–40% reductions in regional synaptic density in AD and non-AD dementias, in proportion to disease severity.19 Postsynaptic pathology can also be measured by PET. For example, TARPγ8 regulates surface expression of postsynaptic AMPA receptors.w159 Ligands targeting TARPγ8 are in human use, including early trials.w160 161 Cerebral protein synthesis rates can be measured with L-[1-11C] Leucine PET,w162 163 a technique that has already been applied to children with developmental delayw164 and young adults with Fragile X syndrome,w165 and holds particular promise for upcoming trials targeting proteostasis and the unfolded protein response.w160
Advances in PET also derive from new methods of analysis. For example, traditional approaches use mass-univariate ‘voxelwise’ tests, or comparisons within specified regions of interest. In contrast, one can study the distribution of binding across disease subtypes. This is particularly powerful where binding affinity varies according to molecular pathology, such as in AV-1451’s affinity with different mutations of the Tau gene in FTD,w129 154 or neuropathological subtypes of FTD.4 8 15 w166 Differences in affinity undermine the contrast between groups for any given region or voxel, but they do not prevent the multivariate approach assessing the similarity of ligand distributions, and classifying individuals into groups based on measures of clustering. This is analogous to the multivoxel pattern analysis techniques used for cognitive decoding in functional MRI (fMRI)20 and magnetoencephalography (MEG).w167
Molecular imaging is no longer solely the preserve of PET. Higher MRI field strengths open up the possibility of richer applications of spectroscopy, with signal to noise ratios sufficient for measurement of GABA in patients.21 Quantitative susceptibility mappingw168 169 assesses the regional burden of paramagnetic substances, with iron particularly linked to cognition in both Alzheimer’s22 and Parkinson’sw170 disease (PD).
Neurodegenerative diseases are often accompanied by vascular comorbidity, with which they can interact.w171 7T high-resolution time-of-flight angiography enables the classification of individual hippocampal vascularisation patterns, and support individual assessments of hippocampal vascular reserve.23 w172 Or, enlarged perivascular spaces may indicate a failure to clear fluid and waste products,w173 including amyloid and tau.w174–176 By combining these novel metrics with classical quantifications like white matter intensities and cortical microbleeds, a comprehensive vascular profile can complement molecular, structural and functional imaging for precision medicine.
Multimodal imaging to test disease mechanisms
Multimodal studies combining PET ligands for metabolism, protein aggregation and neuroinflammation are particularly powerful to examine underlying disease processes. Early studies established the strategy of amyloid PET to confirm underlying Alzheimer’s pathology before assessing neuroinflammation.w152 177 The multimodal approach also reveals associations between neuroinflammation and metabolic impairment,w178–180 and the colocalisation of neuroinflammation and protein aggregation in AD,24 PSPw181 and FTD.8 Such combinations can address the relative prognostic value of imaging markers, and clarify the functionally relevant processes to prioritise for disease modifying treatment.25 Together these studies demonstrate the interplay between critical disease processes, elucidating the cascade of pathogenic mechanisms.
Molecular imaging can be combined with imaging of neuronal connectivity, including diffusion MRI, fMRI and MEG. Neuronal populations are functionally and structurally connected at a number of scales, from microcircuits within a cortical columnw182 through local, modular connectivity communitiesw183 to whole-brain networks.w184
While there is a high degree of correspondence between structure and function in the healthy brain, the same is not necessarily true in dementia. For example, early synaptic loss and neurotransmitter deficits can alter function without cell death (atrophy).26 w185 The transition from presymptomatic to symptomatic stages of neurodegeneration is more closely related to a loss of functional connectivity and information transfer in brain networks than a sudden change in structure.27 In AD, this is reflected in close associations between tau burden and hippocampal function, irrespective of hippocampal volume,28 and a stronger relationship between functional connectivity and memory than with atrophy.w186 Functional adaption may occur in structurally healthy brain remote from the site of neurodegeneration,29 or within areas of early neurodegeneration.w187
Seemingly inconsistent reports of the relationship between atrophy and hypometabolism are reconciled when considered in terms of networks. For example, while published neuroimaging studies of dementia in PD do not show consistent effects when meta-analysed with traditional univariate methods, a network mapping approach revealed consistent dysfunction in a network centred on the hippocampus.30 Similar approaches have highlighted the involvement of different connectivity networks across neurodegenerative syndromes.31
It has been consistently observed in both structural32 and functionalw188 ‘connectomes’, that brain regions that are most densely connected are most vulnerable to neurodegeneration. The properties of these densely connected ‘hubs’w189 can be quantified mathematically with graph theoryw190 or structural equation modelling.w191 The connectomic approach has yielded novel insights into disease mechanisms in dementia, including vulnerability of long connections in Huntington’s disease;w192 initiation of Alzheimer’s33 and mechanisms of hallucinations in Parkinson’s dementia.34
In AD, hub regions may be vulnerable because they are most likely to receive pathological proteins that propagate transneuronally, in a ‘prion-like’ manner.w193–195 Much as countries with highly connected airports are more vulnerable to viral epidemics, brain hubs are more likely to receive pathology from ‘seed’ regions.35–37 w196 197 Combining PET of protein aggregation and functional imaging of brain connectivity using MRI or MEG provides in vivo evidence for this process in humans that was only previously available in animal models,9 w198–201 probing disease across temporal and spatial dimensions.
Although implicated in neurodegenerative disease, transneuronal spread may not be the only cause of hub vulnerability. Multimodal imaging studies demonstrate that in PD, differential gene expression contributesw202 while, in PSP, higher metabolic demand38 or reduced trophic supportw203 may link protein aggregation and abnormal connectivity.9 It is likely that differences in the ultrastructure of the pathological protein affect its propensity to traffic transneuronally,w125 leading to differences between widespread pathology in network-level diseases such as AD and motor neuron disease (amyotrophic lateral sclerosis) vs more focal neurodegeneration in PSP and semantic dementia. Similar principles apply to synucleopathies, with different strains in MSA and LBD.w204
Relating neuronal properties to complex behaviours
Functional imaging can assess real-time connectivity between regions, with fMRI, M/EEG or direct electrode recordings. These methods fall into two broad categories39: (1) ‘functional connectivity’ describes activity between brain regions that is correlated over time or phase coherent, reprising Hebb’s principle that neurons that are wired together fire together and (2) ‘effective connectivity’ refers to directional influences with one region causing change in another; assessed by techniques such as dynamic causal modellingw205 or Granger causality.w206
In the healthy brain, the structure of connections is closely matched to the strength of their functional connectivity, set within overall cortical connectivity gradients40 over a range of spatial scales.w207 This applies at rest (the so-called ‘resting state’) and during tasks.w208 209 However, the structure-function homology breaks down in dementia. The strength of resting-state connectivity between two brain regions typically falls as they are affected by neurodegeneration.41 However, when those regions are engaged by a task, their connectivity may paradoxically increase, perhaps in compensation as they ‘work harder’ to perform a cognitive operation with a less efficient neural architecture.42 Changes in structure-function coupling can also be measured using an approach that considers the gradients of hierarchical organisational,40 which then show decoupling in neurodegenerative diseases such as Parkinson’s.w212
To explain how biological, psychological and social variables contribute to the overall expression of disease, Watershed modelsw213 use the analogy of a river. Intermediate stages of the hierarchy are termed ‘endophenotypes’, which can be observed by cognitive or neural properties. Such modelling of multi-modal neuroimaging can explain complex traits,43 and give insights into the factors that mediate epidemiological observations; for the association between mid-life obesity and late-life neurodegeneration is accounted for by changes in white matter integrity.w214 These models can be extended by the addition of a phenotypic ‘delta’ at the end of the metaphorical river; recognising that multiple cognitive and behavioural outputs stem from different admixtures of common neuronal quantities. Multivariate analysis of large cohortsw215 can relate imaging and cognitive parameters, and describe how this relationship changes with age.w216 217
The way in which age and disease moderates the relationship between structure, function and behaviour is not linear. For example, in MCI, hippocampal or entorhinal atrophy results in hyperactivation of medial temporal lobe circuitry.w186 218 Such local hyperactivity may act in a vicious circle to promote local amyloid deposition.44 This may be a more general property of neurodegenerative disease, as highly active ‘hubs’ initially compensate for declines in structural connectivity by increasing their firing rate.w219 Later in disease this compensation breaks down.45 Cross-sectional studies relating neuronal connectivity to pathological protein deposition have been crucial to corroborating these hypotheses,9 and will be strengthened in future by large, longitudinal cohort studies employing multimodal imaging, allowing generation and testing of prevention and treatment hypotheses.
Assessing microcircuit-levels in humans
So far, we have discussed the macroscopic level of brain structure and function, representing large neuronal populations and the major white-matter connections between them. We turn next to the mesoscopic- and microscopic levels, where initiating events in the neurodegenerative cascade occur.w61–63
Ultrahigh resolution structural imaging with 7-Tesla MRI is sensitive to functional and volumetric changes on the order of hundreds of microns (cf. a grain of sand). However, these field strengths are particularly sensitive to susceptibility changes, and as a result are able to detect microbleedsw220 and iron-dense amyloid plaques that are invisible with standard hospital imaging.w221 Recent advances in laminar fMRI have opened up the ability to examine the functional consequences of degenerative changes within specific cortical layers.46 w222–224
Synapse loss occurs early in AD,w225 perhaps due to direct synaptotoxicity of beta-amyloid and tau aggregates.26 w226–229 This is reflected in functional imaging, for example by tau-related reductions in fMRI hippocampal novelty responses,28 amyloid-related resting state alterations,w230 and inflammation related changed in connectivity of the medial temporal lobe.w231 Amyloid pathology can also be associated with intrinsic neuronal hyperexcitabilityw232 and inhibitory dysfunction.47 w233
The temporal resolution of MEG and EEG (electroencephalography) provide an opportunity to assess the neurophysiological signatures of neurodegenerative diseases, especially in terms of oscillatory dynamics. Neurodegenerative diseases are commonly classified by location of pathology, which tends to track phenotype, but they also cause distinctive changes in the temporal structure of neuronal communication, both at rest,48 and when engaged in a task.w234 These changes can support disease classification even between patients in whom the localisation of pathology is similar, providing a first step towards precision medicine when therapies become available for specific proteinopathies.
Reaching for the microscopic scale with neuroimaging is a challenge, but detailed biophysical models can now be built, to examine the mechanisms of neurodegeneration. For example, dynamic causal modellingw235 can be applied to MEG/EEGw236 and fMRIw205 data, to infer the state of laminar-specific cellular populations and synaptic dynamics.w182 237 When combined with pharmacological intervention, these techniques can assess dynamics that are specific to individual neurotransmitters and receptor populations, allowing a precision of in vivo therapeutic assessment that was hitherto only possible with preclinical models of dementia.
Experimental medicine studies
Neuroimaging can enhance experimental medicine studies in several ways.
First, imaging biomarkers can identify presymptomatic cases. Early intervention may be more effective to reduce the long-term burden of disease, or even prevent symptom onset. For example, amyloid PET imaging is commonly used to identify those with Alzheimer pathology, either as latent disease in presymptomatic individuals, or to confirm AD pathology in MCI.w238 239 There is a 3% annual risk of conversion to amyloid positivity in cognitively normal people over 65, increasing to 7% if positive for apolipoprotein ε4.w240 These individuals show more rapid cognitive decline than amyloid-negative peers.w241 This can enrich clinical trials. However, screening for Alzheimer’s pathology is problematic: elderly amyloid-positive cognitively healthy individuals still only have an 11% annual risk of conversion to MCI or AD,w242 meaning that trials enriched in this way could still take years. Tau imaging with flortaucipir may stratify such cohorts further. Moving forwards, such imaging biomarkers will be increasingly important to identify presymptomatic pathology in those at risk of sporadic or genetically determined dementias, whether AD7 w72 or FTD.5 6
Second, neuroimaging provides an array of surrogate outcome measures that can change more quickly and be quantified more precisely than behavioural and cognitive measures. While rescuing imaging biomarkers is not sufficient, it can provide evidence of a treatment’s effect on the brain as a prelude to clinical endpoints in later phase trials.w243 Atrophy is most widely used in this way, the logic being that a treatment that slows atrophy has influenced neuronal survival, and is more likely to be clinically effective.w244 245 Ultrahigh field imaging (7-Tesla MRI) increases the anatomical resolution and neurochemical sensitivity of MRI, 3–5 x compared with 3T MRI.w246 Hippocampal subfields can be imaged with a resolution of a few hundred microns.w247
However, atrophy resulting from extensive cell death is a late process in the pathogenesis of dementias. Upstream events may be more suitable for earlier intervention, such as the loss of synapses18 19 and inflammation.8 24 To test the relative performance of imaging biomarkers, and against fluidic biomarkers and clinical rating scales, requires a head-to-head comparison. For this, the Deep and Frequent Phenotyping studyw248 is underway, to compare established and novel metrics of the progression of Alzheimer pathology. fMRI, multiligand PET and MEG are longitudinally assessed alongside behavioural measures, CSF, blood and saliva biomarkers.
Third, neuroimaging can test candidates for restoration of the neural mechanisms of aberrant behaviour in small-n studies, over short timescales. For example, MEG demonstrates that frontal lobe neurophysiological signatures of behavioural inhibition are reduced in behavioural variant FTD and partially recovered by correction of the serotonergic deficit in FTD.49 w249–250 No change was demonstrated in behaviour, either because of power or because, using the watershed analogy described above, serotonergic deficiency is only one tributary to the river of behavioural disinhibition in FTD, alongside atrophy, loss of frontal oscillatory connectivity48 w234 and GABA-ergic depletion.21 50
Fourth, by identifying causes of heterogeneity, neuroimaging methods enable cohort enrichment and stratification at inclusion; and can provide post hoc explanations of variation in a treatment response. For example, the selective norepinephrine reuptake inhibitor atomoxetine was unsuccessful in rescuing response inhibition in PD, but subgroup analysis revealed that the drug was effective in those with more severe disease, and intact fronto-striatal connectivity.w251 Preserved cortical outflow tracts were proposed as necessary for behavioural function to be improved following functional restoration ‘upstream’ in prefrontal cortex.w252 253 Such analyses of multivariate data can reveal effects within heterogeneous populations, and develop protocols to predict individual responses to medication, to realise personalised medicine.51 The heterogeneity of a cohort can be formally dissected according to stage (severity) and/or phenotype. For example, the SuStaIn model uses machine learning to distinguish subtypes from progression of disease, against which treatment efficacy could be individually assessed.52
Fifth, neuroimaging with novel PET ligands can be applied in proof of concept and dose finding. For example, inhibition of O-GlcNAcase reduces phosphorylation of Tau in mice,w254 and has been proposed as a therapeutic strategy in AD and PSP. ASN120290 is a novel, orally delivered inhibitor of this enzyme that has completed phase 1 safety trials.w255 In advance of a phase 2 trial, a dose-finding study uses PET to image enzymatic function in vivo.w256 This novel approach may reduce the number of participants needed in phase 2 trials, a crucial advantage when dealing with rare diseases. Similarly, L-[1-11C]-Leucine PET allows in vivo quantification of modulation of the unfolded protein response in upcoming trials.w160
Finally, neuroimaging can add value to clinical trials of investigational medicines, to inform our understanding of disease processes, establishing a ‘positive feedback’ loop in translational research. Early and late phase studies can be designed in a way that includes longitudinal follow-up of large disease cohorts with both established and novel biomarkers.7 The benefit of these approaches is increased by data sharing.
Big data and artificial intelligence
As computing capabilities expand, the concept of ‘big data’ evolves, but it generally describes information too large or complex to be analysed in traditional ways. Artificial intelligence (AI) and machine learning techniques applied to large, multimodal datasets hold promise for the discovery of complex, non-linear relationships between pathological and environmental factors, improving diagnosis, prevention and treatment. However, with larger-scale data one must be careful to minimise the risks of false discovery through statistical chance or hidden biases,53 w257 guard against overinterpretating small effect sizesw258 and maintain interpretability.w259
Big data may arise from a coordinated effort to obtain a standard set of measures, or to repurpose data generated for other purposes such as healthcare. The former is exemplified by the Human Connectome Project, with 4 hours of imaging in 1200 young volunteers,w260 and similar acquisitions in 1200 older individuals, 1350 teens, 500 babies/toddlers and 1500 fetuses.w261 A second example is the UK Biobank, which aims to acquire MRI in 100 000 individuals for combination with lifestyle, biomarker and genomic data.54 Around 40 000 scans have been acquired to date.
Big data can also be built by fusion of smaller studies. Data sharing is now readily available, facilitated by infrastructure like the Dementias Platform UK Portal,w262 enabling researchers to deposit and access imaging and behavioural measures at scale. This facilitates replication, and generation of very large-cross-cohort analyses. Consensus data formats such as brain imaging data structure, initially developed for MRIw263 and now extended to MEGw264 make this process easier. Consortium efforts accounting for differences between scanner and acquisition protocols such as ENIGMA are increasing our ability to pool inference across cohorts, allowing meta-analysis and repurposing.w265
Big data hold particular promise for hypothesis generation, linked to bespoke studies for hypothesis testing. This reduces the risk of recruitment bias resulting in very large normative cohorts. For example, one must consider whether those with predementia are equally likely to volunteer for a cohort study? Their numbers might either be reduced by latent cognitive difficulties or apathy, or increased by a wish to understand subjective cognitive complaints.w266
The simplest approach to the analysis of big data is the application of a standardised univariate method at large scale. However, critical effects may be multivariate, complex and non-linear. Assessing complex effects is better suited to methods collectively known as AI. This umbrella term describes a range of approaches from supervised machine learning classification algorithms like support vector machines,w267 through to ‘deep learning’ efforts that apply multiple, increasingly abstract, processing layers.55 Through such brute force associative techniques, deep learning can achieve very high diagnostic accuracies for dementia.w268 However, these accuracies are vulnerable to over-fitting, and may lack insight into which disease mechanisms are driving classification. They may fail if the classifier comes across variants not previously observed.
The application of big data approaches to dementia also raises ethical considerations, beyond those of privacy, data security and governance common to all healthcare datasets.w269 270 First, it may require a change in mindset among clinical specialists, with hard-earned diagnostic acumen, to trust algorithms and methods that rely on interactions and hidden states that are not transparent. Second, the responsibility for inaccurate diagnosis would be unclear, a problem likely to be particularly challenging for rarer dementia syndromes. Third, the social, financial and psychological consequences of making a predementia diagnosis in the absence of disease-modifying treatment are difficult to quantify. The response to these challenges requires consultation beyond the neuroimaging community, maintaining a clear distinction between applications for research and direct patient care.
A roadmap to clinical trials
We are entering an exciting phase in dementia research, moving from observational to therapeutic studies, with enhanced ability to perform mechanistic analysis of human dementia pathogenesis. Many novel targets have been identified and diverse disease-modifying agents are coming to clinical trials. Although the ultimate goal is to prevent cognitive and behavioural decline in a way that is meaningful for patients and families, and with health economic benefits, there is an intermediate stage of experimental medicine that will need to exploit quantitative imaging of neuronal and physiological function (figure 1).

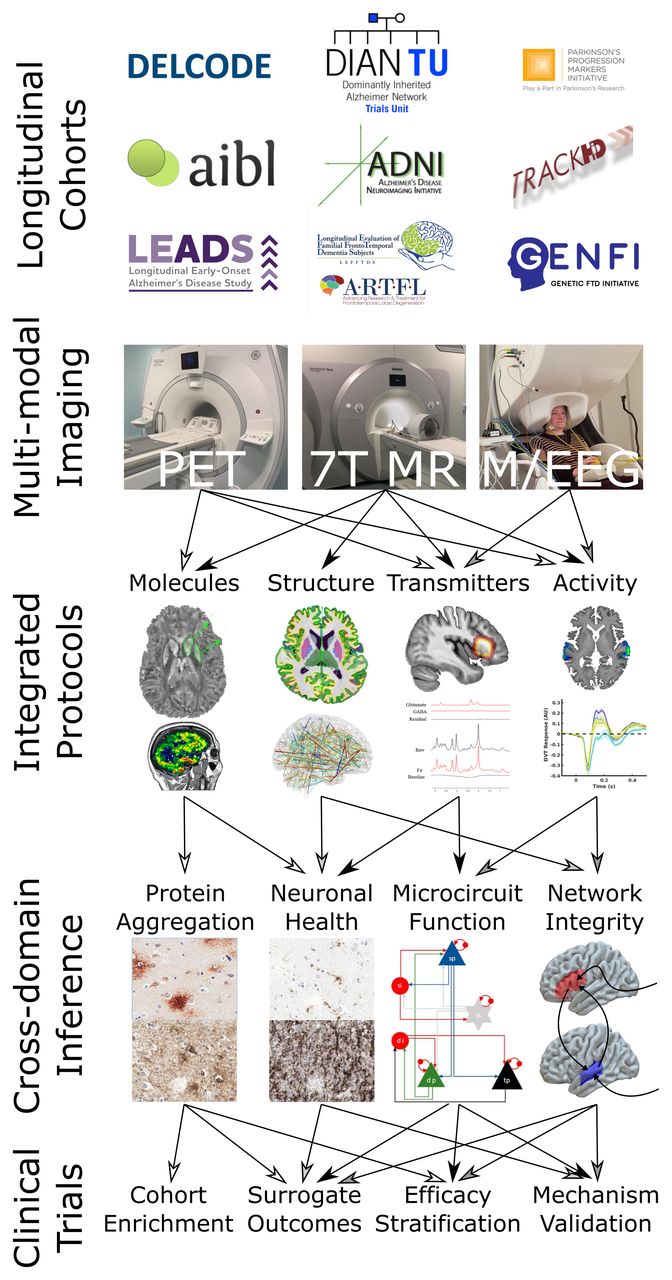{kind=link}
A flow chart illustrating the ways in which carefully designed, multimodal, longitudinal studies can lead to powerful cross-domain inference about neurodegenerative processes. The molecular imaging shown is a 7-tesla QSM (image courtesy of Catarina Rua)w169 and PK-11195 PET.w154 The structural imaging is a FreeSurfer cortical thickness map from a 7-Tesla MP2RAGE image, above a connectivity map that could be derived from diffusion tractography or resting state functional imaging. The transmitter imaging is 7-Tesla MR Spectroscopy (image courtesy of Murley et al).50 The activity images are 7-Tesla auditory fMRI and source-resolved evoked magnetoencephalography responses from superior temporal gyrus. Protein aggregation histopathology images are of amyloid and tau in Alzheimer’s disease. Neuronal health histopathology images are of activated microglia and synaptic integrity. The microcircuit shown is that from an extended dynamic causal model.47 58 The network integrity schematic summarises the inference from coherence and causality techniques for assessing effective connectivity.42 fMRI, Functional MRI; PET, positron emission tomography; QSM, Quantitative Susceptibility Mapping.
There are large, observational, longitudinal cohort studies with multimodal assessment that serve as foundational data on natural history and biomarkers to inform evidence-based interventional trial design. The AD Neuroimaging Initiative changed the landscape by bringing together clinical, imaging and fluid biomarkers to feed into clinical trials. This prototypical study is now in its third phase.56 It sets the stage for many collaborative international studies involving advanced imaging, clinical cognitive and fluid biomarkers, summarised in table 1.
Examples of multicentre dementia studies including an advanced imaging component
As well as their multimodal imaging data, the open science framework of these initiatives encourages collaboration, and replication and validation of findings. At an even larger scale, the ENIGMA consortium spans diseases and facilitates mega-scale imaging/genetic collaborations to answer mechanistic questions only possible with very large numbers of patients.57
Complementary to these initiatives, observational cohorts are critical to deeply phenotype patients, using multimodal imaging and fluid markers. These enable bespoke, experimental and innovative elements to be included and often feed into larger consortia. For example, TRACK-HDw73 and TRACKON-HDw74 are multisite observational cohort studies of Huntington’s disease. Alongside clinical and fluid biomarkers, neuroimaging data include structural and fMRI. Crucially, clinical trials of antisense oligonucleotide designed to reduce mutant huntingtin protein are underwayw276 and neuroimaging metrics refined in observational cohorts inform long term trials of these and similar agents. Similar comprehensive multimodal observational cohorts have been established across neurodegeneration: in PD, the Vision in Parkinson’s study collates retinal, visual, fluid and genetic markers with advanced imagingw277 278 with the aim of refining markers of progression in PD to stratify patients for clinical trials.
This approach emphasises the value of early-stage feasibility trials to produce meaningful insights into disease mechanisms, paving the way for larger trials. For example, the AZA-PD studyw279 is an early phase double-blind placebo-controlled trial of immunosuppression in PD, aiming to modify progression by reducing neuroinflammation. Azathioprine exerts its effect slowly, and it is anticipated that efficacy as measured by traditional motor scores will be weak over 18 months. However, longitudinal PET imaging of neuroinflammation may provide proof of concept for the therapy.
Overall, intelligent design of the neuroimaging component of trials enables true proof of concept, illustrating feasibility and derisking the process by providing early surrogate endpoints that enable Go/Nogo decisions to longer-term cognitive and behavioural endpoints. Medical, scientific, and commercial considerations are complementary, but each enhanced by neuroimaging to work towards a cure for dementia. There are a series of challenges, potential solutions, limitations and hurdles to this overarching goal, summarised in table 2.
Challenges, possible solutions, limitations and hurdles for neuroimaging to support translational medicine in dementia
Summary
Advances in neuroimaging will facilitate the transition from discovery science and drug discovery through to effective and timely clinical trials for dementia and neurodegeneration. We have illustrated the multifaceted roles of PET, MRI and neurophysiology, linked to disease models and AI analysis methods. Understanding of the causes of heterogeneity can be applied to stratify clinical trials, and to the realisation of precision medicine. Building on cross-sector collaboration and best practices for open science, advanced in brain imaging will enhance good clinical care and accelerate dementia prevention and treatment.
see web appenddix for additional references for online supplemental file 1.
Supplemental material
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @tccambs, @rimonaweil, @CambridgeFTD
Contributors Writing—original draft: TEC. Writing—review and editing: RSW, ED, BCD and JBR. Visualisation: TEC.
Funding TEC is supported by an NIHR Clinical Lectureship. RSW is funded by a Wellcome Trust Clinical Research Career Development Fellowship (201567/Z/16/Z). JBR is supported by the Wellcome Trust (103838) and MRC (SUAG051/G101400).
Competing interests RSW has received speaker fees from GE Healthcare. BCD has received personal fees from Arkuda, Eli Lilly, Merck, Wave Lifesciences, Novartis, Oxford University Press, Cambrige University Press, and Elsevier; grants and personal fees from Biogen; and grants from the National Institute on Aging, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. JBR has performed consultancies with Biogen, UCB, Asceneuron, WAVE, Astex, SVHealth; has received research grants from AZ-Medimmune, Lilly, Janssen; and is an editor at Brain.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.