Article Text

Abstract
Since the discovery of the C9orf72 repeat expansion as the most common genetic cause of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis, it has increasingly been associated with a wider spectrum of phenotypes, including other types of dementia, movement disorders, psychiatric symptoms and slowly progressive FTD. Prompt recognition of patients with C9orf72-associated diseases is essential in light of upcoming clinical trials. The striking clinical heterogeneity associated with C9orf72 repeat expansions remains largely unexplained. In contrast to other repeat expansion disorders, evidence for an effect of repeat length on phenotype is inconclusive. Patients with C9orf72-associated diseases typically have very long repeat expansions, containing hundreds to thousands of GGGGCC-repeats, but smaller expansions might also have clinical significance. The exact threshold at which repeat expansions lead to neurodegeneration is unknown, and discordant cut-offs between laboratories pose a challenge for genetic counselling. Accurate and large-scale measurement of repeat expansions has been severely hindered by technical difficulties in sizing long expansions and by variable repeat lengths across and within tissues. Novel long-read sequencing approaches have produced promising results and open up avenues to further investigate this enthralling repeat expansion, elucidating whether its length, purity, and methylation pattern might modulate clinical features of C9orf72-related diseases.
- frontotemporal dementia
- ALS
- C9ORF72
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
A repeat expansion in C9orf72 is the most common genetic cause of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) worldwide.1 2 Besides typical features of FTD and ALS, the expansion is increasingly associated with a broader range of symptoms, including psychiatric manifestations and parkinsonism, and has also been identified in patients with no known family history for neurodegenerative disease.3 4 Early recognition of C9orf72-related illnesses is becoming increasingly important with the advent of disease-modifying therapeutic trials.
The substantial clinical heterogeneity observed in C9orf72 expansion carriers, in terms of phenotype, age at symptom onset and rate of disease progression, suggests that there are underlying disease modifiers which may serve as prognostic factors as well as targets for therapeutic interventions. Drawing on evidence from other neurodegenerative repeat expansion disorders, including Huntington’s disease (HD), several spinocerebellar ataxias and myotonic dystrophy,5 an explanation for this heterogeneity has primarily been sought in repeat length. Studies relating C9orf72 repeat length to clinical features have produced inconsistent results, but they have been hampered by technical difficulties in detecting and sizing expansions.6–8
In this review, we present an overview of the clinical spectrum associated with C9orf72 repeat expansions. We then discuss the role of repeat length in relation to the C9orf72 phenotype. Finally, we provide an in-depth examination of the advantages and limitations of current and novel methods to identify and measure repeat expansions.
Genetics and pathophysiology of C9ORF72 repeat expansions
The C9orf72 repeat expansion is located in a non-coding region and consists of an expanded GGGGCC-repeat. Healthy individuals commonly carry less than 30 repeats, whereas those with C9orf72-associated diseases usually have several hundreds to thousands of repeats.1 2 9 The exact function of the C9orf72 protein is unknown, but it is thought to play a role in autophagy and endosomal trafficking. Three possibly coexisting disease mechanisms have been proposed: (1) haploinsufficiency due to reduced expression of C9orf72 from the expanded allele, (2) formation of repeat-containing RNA foci through bidirectional transcription of the expansion and (3) production of aggregation-prone dipeptide repeat (DPR) proteins via repeat-associated non-ATG translation. These mechanisms have in turn been associated with a wide range of downstream cellular defects, such as alterations in stress granules, proteostasis, nucleocytoplasmic and vesicular transport, mitochondrial function and immunity.10 11 Each disease mechanism alone might not be sufficient to cause neurodegeneration, and a synergistic model in which C9orf72 loss-of-function exacerbates gain-of-function mechanisms has been proposed. A better understanding of their relative contributions across various disease stages is essential in order to determine viable treatment targets. Promising therapeutic strategies currently being researched include small molecules and gene-silencing tools that inhibit C9orf72 transcription, antisense oligonucleotides that bind to and inactivate repeat-containing RNA, antibody-based approaches to inhibit accumulation of DPR proteins, and targeting downstream cellular defects.10 11
Pathologically, C9orf72-related diseases are characterised by cytoplasmic aggregates of transactive response DNA-binding protein 43 (TDP-43) and p62-positive neuronal cytoplasmic inclusions containing DPR proteins.1 2 While TDP-43 pathology coincides neuroanatomically with affected regions of the central nervous system (CNS) in ALS and FTD, DPR proteins generally do not. Sense and antisense C9orf72 RNA foci are widely distributed throughout the CNS in affected expansion carriers.10
Epidemiology of C9ORF72 repeat expansions
A C9orf72 expansion is identified in approximately 5%–10% of all patients with FTD or ALS, and in up to 30% of patients with both diseases.4 12 Collectively, these diseases are referred to as c9FTD/ALS. The prevalence varies strongly between ethnic groups: the highest rates are found in Caucasians and the lowest in Asians.12 The expansion is highly penetrant, with cumulative percentages of 90.9%–99.5% by the age of 83. The first symptoms usually manifest themselves in the fifth decade, but age at onset is highly variable, even within families, and can range from 20 to 90 years.9 13 14 The initial presentation and disease course also vary widely (box 1, table 1); survival after symptom onset ranges from 2 months in rapidly progressive ALS to over 30 years in slowly progressive behavioural variant FTD (bvFTD).14
Common clinical presentations of C9orf72 repeat expansion carriers
Representative case histories of notable C9orf72 expansion carriers, highlighting the variability in clinical presentation, age at symptom onset and disease course.
Case 1
A middle-aged woman was admitted to the psychiatric ward due to severe vital depression and attempted suicide. She had experienced a similar episode of depression with psychosis 2 years previously which was unresponsive to antidepressants and eventually treated with electroconvulsive therapy (ECT). Before that time, she had no history of psychiatric disease. Her mother had exhibited strange social behaviour from her late sixties; her maternal grandfather had been diagnosed with Alzheimer’s disease. Extensive laboratory testing and brain MR imaging were normal; a fluorodeoxyglucose positron emission tomography (FDG-PET) scan revealed bilateral temporal and mesiofrontal hypometabolism compatible with FTD. Her depression was treated successfully with ECT again, but she subsequently suffered progressive cognitive decline and was admitted to a nursing home 5 years after her first episode of depression.
Cases 2 and 3
A man in his thirties presented with a 2-year history of word finding difficulties and memory complaints. Family members also reported mild behavioural changes. His father had been diagnosed with frontotemporal dementia in his seventies and several paternal relatives had suffered from amyotrophic lateral sclerosis (ALS). MRI of the brain demonstrated mild bilateral frontotemporal atrophy, and neuropsychological assessment confirmed deficits in language and executive functioning domains. Very slow progression occurred over several years of follow-up, with behavioural abnormalities such as social disinhibition and loss of decorum becoming more noticeable. Currently, 10 years after the initial presentation, he is still living independently.
His brother presented in his forties with progressive difficulties speaking and swallowing since 3 months. He had subsequently developed weakness of the left arm and leg. His partner also reported increased emotional lability and memory decline. Neurological examination revealed a bulbar dysarthria, tongue wasting with fasciculations and brisk jaw jerk, as well as mild left-sided hemiparesis. Electromyography was compatible with ALS, neuropsychological testing showed mild impairments in social cognition and executive functioning, and MRI revealed moderate, predominantly left-sided frontotemporal atrophy. His condition deteriorated rapidly and he died within 9 months of symptom onset.
Case 4
A middle-aged man was admitted to the psychiatric ward with a 3-month history of somatic delusions. He had become convinced that there were foreign bodies inside his abdominal organs and refused to eat or drink. There was no history of neurodegenerative diseases on the maternal side of his family; his paternal family history was unknown. Neurological examination at the time did not reveal focal abnormalities except positive primitive reflexes and elevated muscle tone. Extensive laboratory testing, MR and FDG-PET imaging, and electroencephalography were inconclusive. Despite antipsychotic medication, the patient deteriorated rapidly into a state of catatonia with limb myoclonus, cerebellar ataxia and apnoeas and required continuous tube feeding. He remained in a fluctuating mute catatonic state over the following 2 years.
Clinical manifestations of C9ORF72 repeat expansions
Cognitive disorders
Frontotemporal dementia
A C9orf72 expansion is found in 20%–25% of familial and 6%–8% of apparently sporadic FTD cases (c9FTD).4 12 The median age at onset of 57 years is similar to that of FTD without a C9orf72 expansion (non-c9FTD),14 15 but survival after symptom onset is shorter (7 vs 11 years), even in the absence of concomitant ALS. The clinical presentation of c9FTD is usually bvFTD, which is characterised by progressive behavioural and personality changes, prominent executive functioning deficits and relative sparing of other cognitive domains,16 although language variants (non-fluent primary progressive aphasia and semantic dementia) may also occur (table 1).14 15 Up to 30% of c9FTD patients develop concomitant ALS during the course of the disease, but routine screening for ALS symptoms in patients with c9FTD is not commonly performed.15 c9FTD cannot be reliably distinguished on clinical grounds from non-c9FTD. However, c9FTD patients may present with atypical signs and symptoms and frequently do not fulfil current diagnostic criteria for FTD.16 17 First, memory impairment is more common in c9FTD and is often the presenting symptom in older subjects, which can lead to misdiagnosis of Alzheimer’s disease (AD).18 19 Second, psychotic symptoms and bizarre, irrational behaviour are common in c9FTD.15 Third, some patients have a protracted or seemingly non-progressive disease course. Fourth, atrophy patterns in c9FTD are highly variable and can include generalised symmetrical cortical atrophy and atrophy of thalamus and cerebellum (figure 1, table 1).19

T1-weighted MR imaging of four affected C9orf72 expansion carriers, demonstrating the diversity of atrophy patterns that can be encountered. MRI scans were made at the time of the initial presentation; patients were between 55 and 62 years of age. (A) mild generalised cortical atrophy; (B) severe generalised cortical cerebral as well as cerebellar atrophy; (C) predominantly left-sided frontotemporal atrophy; (D) moderate symmetrical parietal atrophy as well as mild symmetrical frontotemporal atrophy.
Benign variant FTD
A slowly progressive variant of bvFTD, with isolated neuropsychiatric symptoms and minimal cognitive deterioration over many years of follow-up, has been described in several C9orf72 expansion carriers.20 Furthermore, a recent meta-analysis showed that 2% of patients with so-called benign variant FTD, in whom neuroimaging abnormalities and disease progression are lacking, had a C9orf72 expansion.21 The identification of expansions in benign variant FTD suggests that it is a neurodegenerative condition in at least a subset and that genetic testing should be considered, especially in those with a positive family history.
AD and other dementias
C9orf72 expansions have been identified in a small percentage (<1%) of clinically diagnosed AD patients in several large cohorts.18 22 Most of these cases can probably be attributed to misdiagnoses, especially in older patients with memory impairment and atypical neuroimaging, and might not reflect a causative association between C9orf72 expansions and AD. Accordingly, autopsy studies of expansion carriers with a clinical diagnosis of AD revealed either isolated FTD pathology, or FTD with concomitant AD pathology,23 24 although more pathological studies are needed to confirm this. Importantly, concomitant AD pathology might modulate the clinical phenotype (ie, more amnestic features) and thus conceal the existence of a C9orf72 expansion. Pure AD pathology without FTD pathology reported in three expansion carriers possibly reflects incomplete penetrance. From a clinical perspective, the presence of C9orf72 expansions in clinical cohorts of AD highlights the need to screen FTD genes in AD patients with a positive family history.22
Genetic screening of large series of Lewy body dementia (DLB) patients did not identify C9orf72 expansion carriers, but occasionally, carriers may present as DLB mimics or harbour DLB copathology.
Motor neuron diseases
A C9orf72 expansion is identified in 20%–30% of familial and 5% of apparently sporadic ALS cases.4 12 The median age at onset (57 years) in c9ALS is similar to that of ALS without a C9orf72 expansion (non-c9ALS).13 14 The clinical phenotype of c9ALS covers the entire ALS spectrum, typically presenting with muscle weakness starting in one segment and spreading throughout the motor system (table 1). A bulbar onset of disease, characterised by early dysphagia and dysarthria, is seen in 30%–40% of c9ALS13 14 and appears to be more common than in non-c9ALS and may explain the shorter overall survival in c9ALS reported in some studies.13 14
Cognitive impairment and dementia, mostly of the bvFTD subtype, are more common in c9ALS than in non-c9ALS.15 Accordingly, c9ALS patients show more extensive atrophy of non-motor areas of the frontal cortex than those with non-c9ALS. Although only 20% of ALS patients fulfil FTD diagnostic criteria, an additional 20% has some degree of cognitive impairment, usually in executive functioning, social cognition or language, and 10% shows behavioural changes. Screening instruments that are specifically designed to detect cognitive and behavioural symptoms in ALS, such as the Edinburgh Cognitive and Behavioural Screen, are increasingly used and may improve tailored patient care.
Other motor neuron diseases, including progressive muscular atrophy and primary lateral sclerosis, are rare in C9orf72 expansion carriers.14
Psychiatric manifestations
Psychiatric symptoms occur at a much higher rate across the spectrum of C9orf72-related diseases than in non-c9FTD/ALS. Psychotic features are reported in 20%–60% of c9FTD cases15 17 25 and include delusions and hallucinations in all sensory modalities, although somatic delusions are relatively common.15 Other psychiatric manifestations include mood disorders, obsessive compulsive disorder and catatonia (table 1).25–27 Psychiatric symptoms are occasionally the initial presentation and sometimes occur several years before the emergence of more typical FTD or ALS symptoms.15 27 Misdiagnosis of a primary psychiatric disorder such as late-onset schizophrenia or late-onset bipolar disorder is not uncommon, especially in the case of (near-)normal neuroimaging.27
C9orf72 expansions in primary psychiatric disorders are rare, with prevalence rates below 0.2% in several large cohort studies.28 29
Movement disorders
Parkinsonism
Concomitant parkinsonism occurs more frequently in c9FTD/ALS than in non-c9FTD/ALS, and is especially common in bvFTD patients, with up to 75% developing a variable degree of parkinsonism during the disease course.30 Symptoms include symmetrical or asymmetrical bradykinesia, rigidity, falls and gaze palsy, often with little or no tremor.30–32
An initial presentation of parkinsonism, ataxia or apraxia has probably led to an erroneous diagnosis of Parkinson’s disease (PD), progressive supranuclear palsy, corticobasal syndrome or multisystem atrophy in several expansion carriers.31 33 Some of these patients lacked typical FTD or ALS signs for several years after presentation.31–33 Parkinsonism in expansion carriers has been related to neuronal loss as well as TDP-43 and p62 pathology reported in the basal ganglia in a few pathology-proven studies.34 35 Accordingly, while some cohorts of clinically diagnosed PD report occasional (<1%) expansions,33 36 across two large studies of autopsy-proven PD (pooled sample size >800 patients), just one expansion carrier was found who had both typical PD pathology as well as C9orf72-mediated pathology.34 37
HD-like syndrome
A C9orf72 expansion has been identified in up to 5% of patients with HD-like (HDL) syndrome, which is clinically indistinguishable from HD but without the typical CAG repeat expansion in the HTT gene, making it the most common genetic cause of HDL-syndromes.38 39 There is no evidence for a causal relationship between C9orf72 and HD, but these findings are relevant for clinical practice as they demonstrate that expansion carriers may present with atypical signs including chorea, dystonia, tremor, rigidity, bradykinesia and myoclonus.39 Neuropathological data is needed to determine whether C9orf72-linked pathology in HD-related brain regions might underlie these unusual presentations. Remarkably, repeat expansions in HTT were recently identified in a small number of non-c9FTD/ALS patients, supporting a phenotypical and possibly etiological overlap between HD and FTD/ALS.
Other movement disorders
Other movement disorders in expansion carriers include cerebellar ataxia and a case with a clinical diagnosis of Creutzfeldt-Jakob disease.3 12 These cases probably represent misdiagnoses of unusual presentations of c9FTD/ALS and further highlight the diversity of the clinical spectrum.
Recommendations for genetic testing
Offering genetic testing for a C9orf72 repeat expansion is generally recommended in patients with concomitant FTD/ALS or familial FTD or ALS, but the best approach for seemingly sporadic FTD or ALS is still under debate.40 This is exemplified by two surveys published in 2017 which found that genetic testing is offered to 90% of familial ALS patients versus just 30%–50% of seemingly sporadic cases. The relatively frequent identification of expansions among seemingly sporadic cases, coupled with better availability of genetic testing and the promise of gene-targeted therapy, has led to an increasing tendency to consider genetic testing in all FTD (especially bvFTD) and ALS patients, regardless of family history.40 41 Importantly, although certain clinical features may prompt the clinician to suspect an underlying C9orf72 expansion (table 1), many c9FTD/ALS patients do not show distinctive features; relying on such features to select patients for genetic testing therefore carries the risk of missing expansion carriers.
While the overall frequency of a C9orf72 repeat expansion in psychiatric disease is very low, genetic testing should be considered in late-onset (>40 years) cases with cognitive decline, prominent cerebral or cerebellar atrophy, or a family history of FTD, ALS or psychiatric disease (table 1).41 Offering genetic testing has also been recommended for patients presenting with parkinsonism and concomitant upper or lower motor neuron symptoms or early neuropsychiatric or cognitive impairment.32
All patients and their families should receive genetic counselling prior to genetic testing, emphasising limitations of current testing methods and potential implications of test results.40 42 In many situations, coupling C9orf72 repeat expansion assays with multigene sequencing panels, which screen for the most common genetic mutations causing neurodegenerative disorders, is advisable considering the substantial overlap in symptomatology between various mutations and the risk of mutations in multiple disease-causing genes .40
Clinical significance of repeat length
Normal and pathological repeat length
There is no clear consensus on which C9orf72 repeat length is pathogenic. Most laboratories consider a repeat length of less than 20–30 in blood to be normal, while more than 200 repeats are very likely pathogenic.42 Small expansions (ie, up to 200 repeats) represent a grey area in which some carriers develop symptoms, while others do not.43–45 The interpretation of small expansions presents a major challenge to clinical genetic counselling and can have far-reaching consequences for affected families.42
Repeat length variation across and within tissues
Measuring C9orf72 repeat length, determining the threshold for pathogenic repeat length and identifying possible associations with clinical features is greatly complicated by instability of the expansion, which results in different repeat lengths across and within tissues of the same individual.3 8 43 44 46–48 Furthermore, expansion sizes in blood might not accurately reflect sizes in the CNS and should be interpreted cautiously.8 47 Direct comparisons of blood and brain tissue from the same individual demonstrate similar sizes in some subjects, but completely different sizes in others.8 47 48 This is illustrated by several cases with small expansions in blood and long expansions in brain tissue.8 43 45 48 The interpretability of repeat lengths is also hampered by somatic instability in blood, resulting in big smears spanning a range of expansion sizes and possibly extra bands.3 47 49
In cell lines, expansion sizes should also be interpreted carefully. Cell lines usually contain fairly small expansions as compared with patient tissues.7 8 Additionally, due to their oligoclonal nature, they may demonstrate multiple concise bands, as shown for fibroblasts, lymphoblastoid cells, induced pluripotent stem cells (iPSCs), iPSC-derived neurons.7 8 49
The risk of false-negative genetic testing due to somatic instability in individuals with non-expanded alleles in blood is probably limited, since up to 30 repeats appear to be stable across tissues.43 48 Accordingly, no expansions were found in neural tissue of a small number of ALS patients with non-expanded alleles in blood.48 50
Clinico-pathological associations with expansion sizes
Although significant associations between clinical features and expansion size have been reported, findings are inconsistent, which might, to some extent, be explained by the substantial differences between tissues and subjects investigated. Some studies detected differences in expansion size between c9FTD and c9ALS,46 49 but others could not replicate these findings.3 8 48
Potential associations have been detected between repeat length in various tissues and age at symptom onset.3 7 8 47–49 51 For example, one study reported a positive association between repeat length in the frontal cortex and age at onset in c9FTD.8 At the same time, the C9orf72 expansion might further expand with increasing age.8 47 49 Age at sample collection and age at symptom onset are generally highly correlated, making it unclear whether longer expansions actually delay symptom onset or simply reflect increased repeat length as an individual ages. Since smaller repeat lengths have been observed in young presymptomatic expansion carriers,47 49 the latter explanation seems plausible. Interestingly, associations with survival after onset suggest that smaller expansions might be beneficial.8 48 49
To replicate these associations, in-depth studies are necessary, taking into consideration differences between tissues, disease subgroups (c9FTD and/or c9ALS), age at collection and disease status (affected or unaffected). These studies could also reveal associations with pathological C9orf72-mediated features, such as levels of C9orf72 transcripts, RNA foci and DPR proteins.
Anticipation
A repeat expansion might lengthen in successive generations, which could lead to an earlier age at onset and/or more severe disease phenotype. This phenomenon—also known as anticipation—has been reported in numerous repeat expansion disorders.5
Interestingly, an increase of roughly 1000 repeats from one generation to the next was described in a c9FTD family,44 suggestive of anticipation. Moreover, in a subset of families, an earlier age at onset was observed in younger generations.1 52 Because Southern blots were not performed in most parent-offspring pairs included in these studies, it remains unclear whether or not reported differences in age at onset can be attributed to an increase in expansion size. One could postulate that individuals with a family history of FTD or ALS may seek medical attention sooner. Furthermore, a growing awareness of FTD and ALS in the general population and among clinicians as well as the availability of unified diagnostic criteria may lead to an earlier diagnosis. Besides, these findings could be (partially) driven by selection and/or recall bias.
Importantly, two recent studies actually provided evidence against anticipation in C9orf72-related diseases.47 51 These studies detected a reduction of the expansion size in successive generations, accounting for more than 50% of transmissions.47 51 Intriguingly, contractions were frequently encountered in paternal transmissions, but rarely in maternal transmissions.47 It should be noted that both studies were based on repeat lengths in blood which may not correlate with those in the brain. This is exemplified by a case report of an unaffected father with approximately 70 repeats in blood, while his children harboured long expansions.53 Although this could indicate a jump from a premutation to a disease-causing expansion in the next generation,53 postmortem tissue of the father later revealed long expansions in his brain,45 stressing the importance of determining expansion sizes in brain tissue. As such, to unravel whether anticipation contributes to C9orf72-linked diseases, large-scale studies focusing on brain tissue from multiple generations are needed.
Detecting and sizing C9ORF72 repeat expansions
Detection of C9orf72 repeat expansions with PCR-based approaches
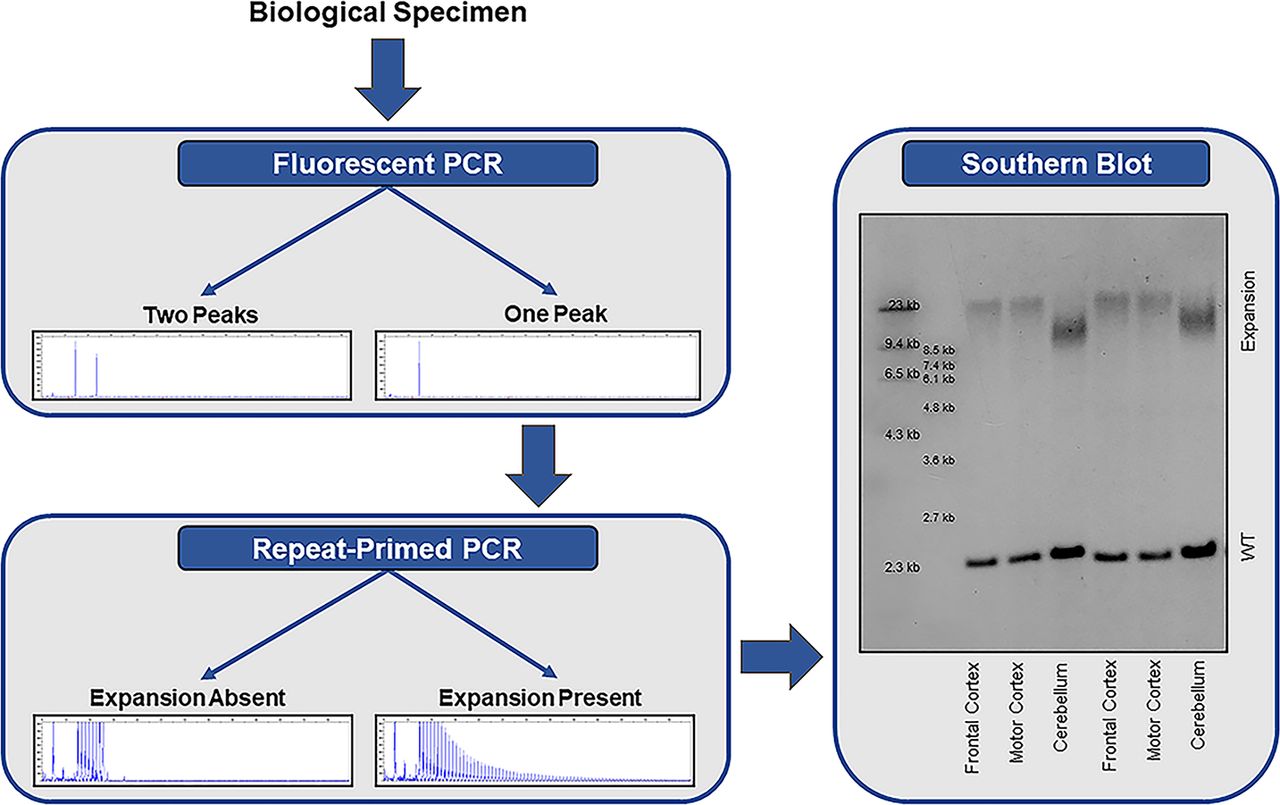
The most commonly applied methods for detecting repeat expansions are PCR-based approaches; a two-step protocol consisting of a fluorescent PCR fragment-length analysis followed by repeat-primed PCR is recommended (figure 2).1 A fluorescent PCR can specify the size of alleles within the wild-type range. If an individual has two alleles with a different number of repeats (eg, two repeats and five repeats), then two peaks are detected and no additional tests are required. If, however, a single peak is observed, then a repeat-primed PCR is warranted. A repeat-primed PCR is able to determine whether an apparently homozygous individual carries two wild-type alleles with exactly the same number of repeats, or whether a wild-type allele is present in addition to an expanded allele that is too long for detection using a fluorescent PCR, in which case a characteristic stutter pattern is seen. Reactions should be repeated (eg, with more DNA), when results are difficult to interpret. If the fluorescent PCR repeatedly fails (ie, does not reveal any peaks) in the presence of a stutter pattern on the repeat-primed PCR, then a homozygous expansion should be considered.

{kind=link}
{kind=link}
Stepwise approach for the detection of a C9orf72 repeat expansion. Fluorescent PCR fragment-length analysis is used to determine the number of alleles in the wild-type range. If a single peak is observed, then repeat-primed PCR is warranted to determine whether the individual carries two wild-type (WT) alleles with the exact same number of repeats, or whether a repeat expansion is present that is too long for detection by fluorescent PCR. if a repeat expansion is present, a characteristic stutter pattern is observed on repeat-primed PCR. especially in a clinical setting, it is recommended to confirm the presence of an expansion using southern blotting.
Importantly, an international study demonstrated that 6 out of 14 participating laboratories misclassified at least one sample when using a repeat-primed PCR alone.6 Given that the accuracy increased when both a fluorescent PCR and repeat-primed PCR were used,6 a two-step protocol is the preferred method to determine the presence or absence of a C9orf72 repeat expansion in a research setting. A subset of misclassifications should probably be attributed to the genomic complexity of the C9orf72 region. Sequence variants (eg, deletions and insertions) have been described downstream of the expansion.9 54 Depending on how the repeat-primed PCR has been designed and whether it has been thoroughly optimised, the presence of sequence variants might hamper the amplification of the expansion, and therefore, lead to a false negative repeat-primed PCR. Interestingly, it has been suggested that a downstream deletion lengthens the expansion. A 10 bp deletion, for instance, removes several non-GC-bases, extending the GC-rich region with imperfect repeats.9 Although this might be relevant for relatively small expansions, possibly decreasing stability of the repetitive area,9 one could doubt whether the addition of a few extra GC-bases to a long expansion will have a major effect.54 Regardless, these sequence variants should be taken into consideration since they may reduce the accuracy of the repeat-primed PCR
Detection and sizing of expanded C9orf72 repeats with Southern blotting
The gold standard to detect a C9orf72 repeat expansion is a Southern blot (figure 2). Results are highly consistent across laboratories,6 and Southern blotting should be used to confirm the presence of an expanded repeat, especially in a clinical setting. Moreover, Southern blotting can clarify whether an individual is heterozygous or homozygous for the expansion (ie, presence or absence of a band corresponding to the wild-type allele). As opposed to a repeat-primed PCR, Southern blotting is also capable of estimating the size of the expansion.
Various Southern blot protocols have been published to estimate the size of expanded C9orf72 repeats using different restriction enzymes and probes.3 8 46–49 Whether an area close to the expansion is targeted7 8 47 or the expansion itself,3 49 also affects the Southern blot: while probes that target the repeat frequently produce a strong signal, they can hybridise to GGGGCC-repeats in other areas (outside of the C9orf72 locus), and additionally, they may not visualise the wild-type allele, which is often used as an internal control that the Southern blot worked and which helps to elucidate whether someone is homozygous for the expansion.7 Even though Southern blotting can accurately determine the presence or absence of an expanded C9orf72 repeat and estimate its size, it is a time-consuming and technically challenging method requiring high-quality DNA.8
Detection of C9orf72 expansions with short-read sequencing
Several tools have been developed to detect a C9orf72 repeat expansion in PCR-free short-read whole-genome sequencing (WGS) data. The ExpansionHunter tool correctly identified all 212 C9orf72 expansion carriers in a cohort of over 3000 ALS patients.55 Furthermore, a new version of this tool has been recently released that uses sequence graphs to handle complex loci. Despite encouraging results, it remains difficult to reconstruct expansions based on short-read sequencing techniques.55
Employing long-read sequencing to assess sizes, interruptions and methylation patterns
Long-read sequencing techniques are a promising upcoming strategy to evaluate large repeat expansions. Popular platforms have been developed by Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio). ONT’s nanopore technology measures changes in current as DNA passes through a nanopore,56 57 whereas PacBio’s single-molecule real-time technology measures fluorescence when nucleotides are added.56 58 In theory, long-read sequencing can span the entire C9orf72 expansion, which facilitates reliable estimation of expansion sizes. Another major advantage of long-read sequencing is its ability to evaluate sequence content. It is possible, for example, to determine the presence of interruptions in C9orf72 repeat expansions,59 which is highly relevant since interruptions serve as disease modifiers in other repeat expansion disorders.5 Furthermore, long-read sequencing provides the opportunity to examine the methylation status of expanded C9orf72 repeats.60 Interestingly, while hypermethylation of the C9orf72 promoter is well documented, little is known about the methylation status of the expanded repeat itself in c9FTD/ALS patients. It has been suggested that the expansion is methylated; however, quantitative assays have yet to be developed. Importantly, long-read sequencing requires high-molecular-weight genomic DNA, and DNA should be extracted using suitable protocols to ensure that the DNA, and therefore, the expansion, remains intact.
Several long-read sequencing technologies have been tested for C9orf72-linked diseases.59 60 Long-read WGS on ONT’s MinION platform and PacBio’s Sequel platform only yielded a relatively low number of reads covering the expanded allele. Newer CRISPR-Cas9-based enrichment strategies to specifically examine the C9orf72 locus have considerably increased the yield.59 60 These proof-of-concept studies demonstrate the feasibility of long-read sequencing approaches, and further protocol optimisation is expected to improve their accuracy and efficiency, facilitating implementation of long-read sequencing in research and eventually in clinical settings.
Other potential genetic disease modifiers
Besides repeat length, several other potential genetic disease modifiers have been identified, highlighting that a complex interplay of various genetic factors probably contributes to the phenotypic variability of the C9orf72 repeat expansion. For example, intermediate repeat expansions in ATXN2, a known risk factor for ALS, appear to be more common in c9ALS than in c9FTD, suggesting that they might drive the clinical phenotype towards ALS rather than FTD. Similarly, single-nucleotide polymorphisms (SNPs) in TMEM106B appear to protect specifically against c9FTD, but not c9ALS, and two CpG-SNPs (ie, SNPs that affect DNA methylation patterns) at the C6orf10/LOC101929163 locus are associated with age at symptom onset. The rare coexistence of pathogenic mutations in other FTD or ALS-associated genes, including GRN, MAPT, TBK1, TARDBP, SOD1 and FUS might in some cases act synergistically to influence disease severity and/or phenotypic features.
Conclusions and future directions
The C9orf72 expansion is highly clinically heterogeneous, and clinicians need to be aware of the wide spectrum of associated symptoms in order to provide a timely diagnosis and management and to select patients for clinical trials. Despite extensive research, the genetic basis of this heterogeneity remains poorly understood. Novel sequencing techniques that enable thorough assessment of the expansion size as well as its purity and methylation levels may reveal new associations with clinical features. The measurement and interpretation of repeat length is greatly complicated by the presence of somatic mosaicism, and a better understanding of repeat instability is needed to elucidate potential genotype–phenotype correlations as well as to determine the viability of therapeutic approaches aimed at stabilising repeat expansions.
The lack of consensus on clear cut-off values for pathogenicity poses a major challenge for clinical genetic counselling. Further research is also needed to determine whether intermediate repeat lengths have any clinical significance which, remarkably, have been associated with normal to upregulated C9orf72 expression,45 47 in contrast to the downregulation seen in patients with long expansions.
Besides repeat length, several other potential genetic determinants have been identified. Possibly, multiple phenotypic modifiers are present at the same time, concealing clear associations with each individual modifier. Future research into the various modifiers will be essential to predict the expected disease course, which in turn will be valuable both for patient management and for patient stratification in clinical trials.
Supplemental material
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ELvdE and JLJ contributed equally.
MvB and JCVS contributed equally.
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors ELvdE, MvB, HS and JCVS contributed to conception and design of the review and drafting of the manuscript and figures. JLJ and AW contributed to the drafting of the manuscript and figures. All authors critically reviewed the manuscript and approved the final draft.
Funding ELvdE, HS and JCVS are supported by two Memorabel grants from Deltaplan Dementie (The Netherlands Organisation for Health Research and Development and Alzheimer Nederland; grant numbers 7330550813 and 733050103), The Bluefield Project to Cure Frontotemporal Dementia, the Dioraphte foundation (grant number 1402 1300) and the European Joint Programme-Neurodegenerative Disease Research (JPND, PreFrontALS). MvB is supported by the Muscular Dystrophy Association (MDA), ALS Association and National Institutes of Health (NIH) grants R21 NS110994 and R21 NS099631.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.