Article Text

Abstract
Objective To determine clinical manifestations, immunotherapy responsiveness and outcomes of glutamic acid decarboxylase-65 (GAD65) neurological autoimmunity.
Methods We identified 323 Mayo Clinic patients with high-titre (>20 nmol/L in serum) GAD65 antibodies out of 380 514 submitted anti-GAD65 samples (2003–2018). Patients classified as having GAD65 neurological autoimmunity after chart review were analysed to determine disease manifestations, immunotherapy responsiveness and predictors of poor outcome (modified Rankin score >2).
Results On review, 108 patients were classified as not having GAD65 neurological autoimmunity and 3 patients had no more likely alternative diagnoses but atypical presentations (hyperkinetic movement disorders). Of remaining 212 patients with GAD65 neurological autoimmunity, median age at symptom onset was 46 years (range: 5–83 years); 163/212 (77%) were female. Stiff-person spectrum disorders (SPSD) (N=71), cerebellar ataxia (N=55), epilepsy (N=35) and limbic encephalitis (N=7) could occur either in isolation or as part of an overlap syndrome (N=44), and were designated core manifestations. Cognitive impairment (N=38), myelopathy (N=23) and brainstem dysfunction (N=22) were only reported as co-occurring phenomena, and were designated secondary manifestations. Sustained response to immunotherapy ranged from 5/20 (25%) in epilepsy to 32/44 (73%) in SPSD (p=0.002). Complete immunotherapy response occurred in 2/142 (1%). Cerebellar ataxia and serum GAD65 antibody titre >500 nmol/L predicted poor outcome.
Interpretation High-titre GAD65 antibodies were suggestive of, but not pathognomonic for GAD65 neurological autoimmunity, which has discrete core and secondary manifestations. SPSD was most likely to respond to immunotherapy, while epilepsy was least immunotherapy responsive. Complete immunotherapy response was rare. Serum GAD65 antibody titre >500 nmol/L and cerebellar ataxia predicted poor outcome.
Data availability statement
Data are available on reasonable request. Deidentified participant data will be made available to any qualified investigator on reasonable request directed to the corresponding author (NLZ).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Glutamic acid decarboxylase-65 (GAD65) is an enzyme required for synthesis of gamma-aminobutyric acid, a major central nervous system inhibitory neurotransmitter.1 Antibodies targeting GAD65 are a biomarker of type 1 diabetes mellitus (T1DM). Low titres in serum lack clinical specificity for autoimmune neurological disease, and may be detected in patients with alternative neurological diagnoses, isolated T1DM or even healthy controls.2 3 In contrast, high-titre GAD65 antibodies, defined in our laboratory as more than 20 nmol/L in serum (over 1000-fold higher than the upper limit of normal), reportedly confer high clinical specificity for GAD65 neurological autoimmunity.2 GAD65 antibodies appear unlikely to be pathogenic given the intracellular location of GAD65, and may instead be a surrogate marker of cytotoxic T-cell-mediated disease in patients with associated neurological syndromes.1 Stiff-person spectrum disorders (SPSD) were first characterised by Moersch and Woltman in 1956 and later determined to be a prototypical presentation of GAD65 neurological autoimmunity.4–6 Other manifestations of GAD65 neurological autoimmunity that have since been described include cerebellar ataxia, epilepsy, limbic encephalitis (LE), cognitive impairment, myelopathy and/or brainstem dysfunction. These reports, however, are limited by small sample sizes or restriction to individual phenotypes, precluding complete disease characterisation.2 3 6–19 We, herein, evaluate the clinical manifestations, immunotherapy responses and outcomes of a large patient cohort with high-titre GAD65 antibodies who were systematically determined to have GAD65 neurological autoimmunity.
Methods
Patients provided written consent to the use of their records for research.
Identification of patients with GAD65 neurological autoimmunity
We retrospectively identified 323 patients with high-titre GAD65 antibodies (defined as >20 nmol/L in serum based on previous work demonstrating high clinical specificity for GAD65 neurological autoimmunity at this cut-off) detected in the Mayo Clinic Neuroimmunology Laboratory out of 380 514 samples submitted for anti-GAD65 testing from January 2003 to May 2018, using radioimmunoassay (RIA) as previously described.2 Their electronic medical records (EMRs) were then reviewed by two neurologists with fellowship training in Neuroimmunology/Autoimmune Neurology (AB and NLZ). Patients with non-neurological presentations (eg, GAD65 antibody detected as part of T1DM evaluation), as well as those with neurological presentations but a more likely alternative diagnosis than GAD65 neurological autoimmunity, were classified as not having GAD65 neurological autoimmunity; these patients were excluded from study analysis and summarised separately. Patients with no more likely alternative diagnosis but an atypical presentation for GAD65 neurological autoimmunity were also excluded from study analysis but described separately, to ensure potentially novel disease phenotypes were not overlooked. Remaining patients were classified as having GAD65 neurological autoimmunity, and data relating to their clinical presentation, neuroimaging, electrophysiological testing, laboratory findings, immunotherapy responses, and outcomes as measured by modified Rankin score (mRS)20 were extracted from their EMRs for analysis.
Diagnosis of disease manifestations
Diagnosis of disease manifestations in GAD65 neurological autoimmunity was based on clinical assessment by a Mayo Clinic physician with expertise in the disorder of interest, alongside EMR review by AB and NLZ as outlined above to ensure no more likely alternative diagnosis was present. Electrophysiological data (ie, auditory startle reflexes, exteroceptive responses and/or electromyography for SPSD as described previously,7 electroencephalography for epilepsy) were frequently gathered, but an abnormal electrophysiological study was not required for diagnosis given imperfect test clinical specificity.21 22 Patients with SPSD were classified as classical SPSD (trunk and limb involvement), partial SPSD (trunk or limb involvement), or SPSD with prominent exaggerated startle. LE was defined as medial temporal lobe T2-hyperintensity with subacute disease onset of less than 3 months. Cognitive impairment was diagnosed by the treating physician based on the Kokmen short test of mental status23 and/or formal neuropsychometric testing.
Outcome measures
Response to immunotherapy (corticosteroids, intravenous IG, plasma exchange (PLEX), rituximab, cyclophosphamide and/or autologous stem cell transplantation) was classified as no response, partial response, near-complete response (ie, minimal residual clinical signs/symptoms), or complete response (ie, no residual clinical signs/symptoms), as well as sustained (defined as benefit persisting for greater than 3 months) or non-sustained, based on review of the treating Mayo Clinic physician’s documentation by AB and NLZ. A poor outcome was defined as mRS >2 at last clinical follow-up.
Statistical analyses
Statistical analyses were performed using JMP Pro V.14.1.0. Continuous and categorical variables were reported as median (range) and number (percentage), respectively. Differences across multiple groups were assessed by the Kruskal-Wallis, Pearson’s χ2 or Fisher’s exact test for multiple categories, as appropriate. Associations with a poor outcome at last clinical follow-up were explored by univariate logistic regression analysis, while the simultaneous effect of multiple significant variables was assessed by multivariate logistic regression. A two-sided p<0.05 was considered statistically significant. Adjustment for multiple comparisons was not performed.24 The relationships among manifestations of GAD65 neurological autoimmunity were depicted using circular visualisation in R.25
Results
One in three patients with high-titre GAD65 antibodies were classified as not having GAD65 neurological autoimmunity
Of 323 patients with high-titre GAD65 antibodies, 37 (11%) had non-neurological presentations (eg, GAD65 antibody detected as part of T1DM evaluation) and were excluded from study analysis. Seventy-one of 323 patients (22%) were determined to have a more likely alternative diagnosis than GAD65 neurological autoimmunity after review of their EMR; these patients were excluded from study analysis but are summarised separately (online supplemental table 1). Three of 323 patients (1%) without more likely alternative diagnoses but presentations atypical for GAD65 neurological autoimmunity (hyperkinetic movement disorders) were also excluded from study analysis but are described separately (table 2). The remaining 212 of 323 patients (66%) were classified as having GAD65 neurological autoimmunity for study analysis. The median serum anti-GAD65 titre among patients classified as having a more likely alternative diagnosis was significantly lower compared with patients classified as having GAD65 neurological autoimmunity (149 nmol/L vs 534 nmol/L, p<0.0001), and was not significantly different compared with patients with non-neurological presentations (149 nmol/L vs 164 nmol/L, p=0.71). The process of classifying patients as having GAD65 neurological autoimmunity is depicted via flow diagram (figure 1).
Supplemental material
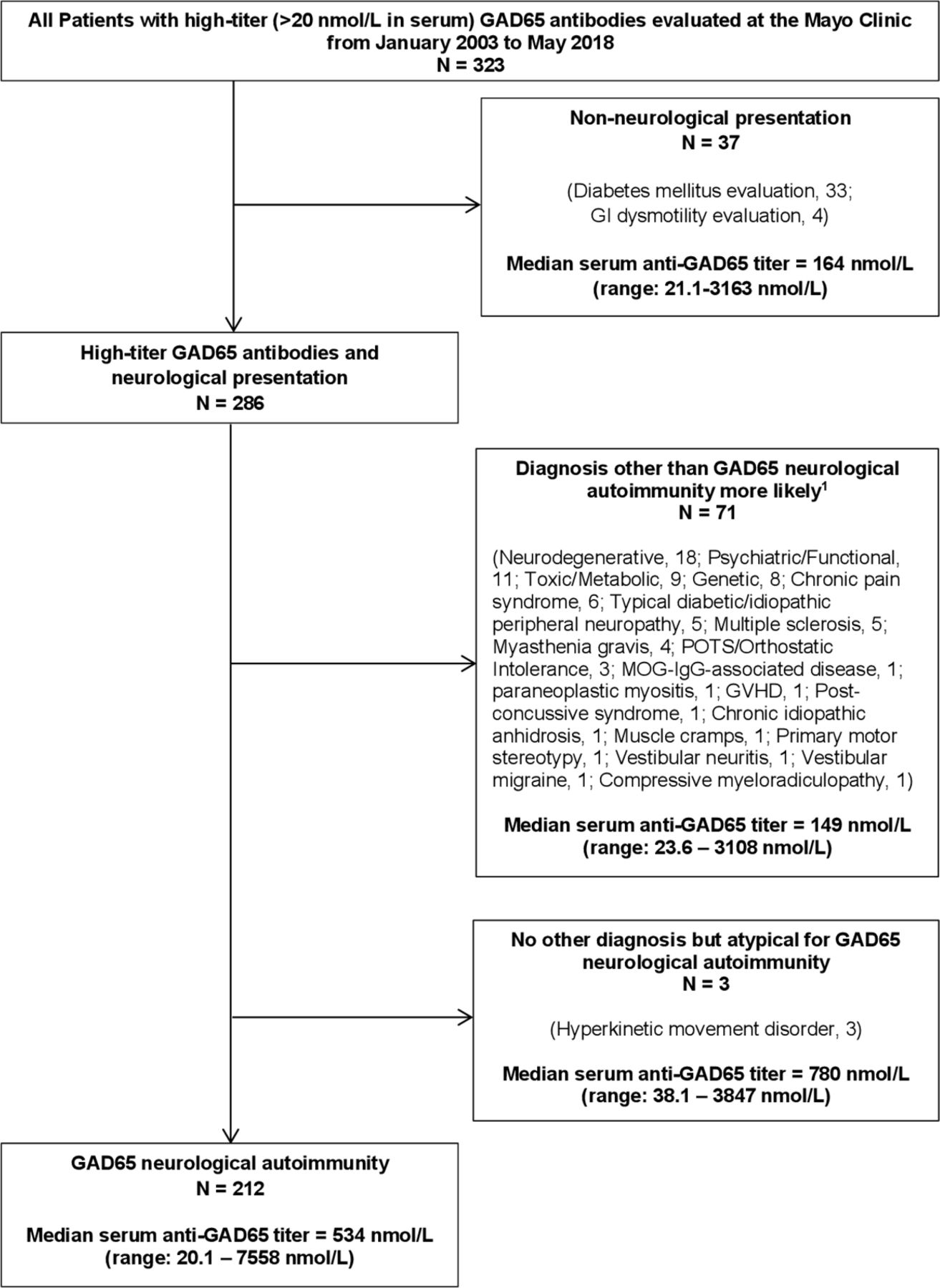
Flow diagram depicting patient selection for study inclusion. GAD65, glutamic acid decarboxylase-65.
Patients with high-titre GAD65 antibodies and hyperkinetic movement disorders
Defining the core and secondary manifestations of GAD65 neurological autoimmunity
Through EMR review, we found that SPSD, cerebellar ataxia, epilepsy without LE (simply referred to hereafter as epilepsy unless otherwise specified) and LE could all occur in isolation. These were thus designated core manifestations of GAD65 neurological autoimmunity. Patients with two or more core disease manifestations were designated overlap syndromes, with the exception of LE and epilepsy (all patients with LE had seizures). No patient had cognitive impairment, myelopathy or brainstem dysfunction reported in isolation (ie, in the absence of SPSD, cerebellar ataxia, epilepsy or LE). These co-occurring phenomena were thus designated secondary manifestations of GAD65 neurological autoimmunity.
Core manifestations of GAD65 neurological autoimmunity are SPSD, cerebellar ataxia, epilepsy and LE
The clinical characteristics, immunological/cancer associations and laboratory results of all 212 patients with GAD65 neurological autoimmunity are presented in table 1. The median age of symptom onset was 46 years (range: 5–83 years) and 163/212 (77%) were female. Concurrent systemic autoimmunity was documented in 125/212 (59%) patients with GAD65 neurological autoimmunity and was most often thyroid disease (72/212, 34%), T1DM (63/212, 30%), and/or pernicious anaemia (40/212, 19%). A diagnosis of cancer within 5 years of symptom onset was reported in 9/212 (4%). Stratification of these findings by core manifestation is included in table 1, and discussed in relevant sections below.
Characteristics of 212 patients with GAD65 neurological autoimmunity*
Stiff-person spectrum disorders
SPSD was the most common core manifestation and was often classical in presentation
The most common core manifestation was SPSD, which was reported in 107/212 (50%). The majority (73/107, 68%) had classical SPSD. Partial SPSD was documented in 30/107 (28%), and a small minority were classified as SPSD with prominent exaggerated startle response (4/107, 4%). Electrophysiological findings supportive of SPSD were reported in 52/70 (74%). Common findings documented on clinical assessment included spasms (93/107, 87%), gait dysfunction attributed to SPSD (85/107, 79%) and hyperlordosis (49/107, 46%).
Cerebellar ataxia
Cerebellar ataxia was the second most common core manifestation and often affected gait
Cerebellar ataxia was reported in 91/212 (43%). Gait ataxia was most frequently documented (76/91, 84%), followed by limb ataxia (63/91, 69%) and ataxic dysarthria (47/91, 52%). On brain MRI, cerebellar atrophy was observed in 24/91 (26%); no patient had cerebellar T2-hyperintensity or gadolinium enhancement indicative of cerebellitis.
Rare paraneoplastic cases associated with cerebellar ataxia
While a diagnosis of cancer within 5 years of symptom onset was reported in only 9/212 (4%), this ranged from 0/71 (0%) in SPSD to 6/55 (11%) in cerebellar ataxia (p=0.01). Cancers diagnosed included thyroid cancer, breast cancer, lung cancer, and thymoma (table 1).
Epilepsy
Epilepsy was classically temporal lobe in origin and occasionally musicogenic
Epilepsy with or without LE was reported in 62/212 (29%). Seizures were focal-onset in 56/62 (90%) and unknown-onset in 6/62 (10%). Seizures most often localised to the medial temporal lobe (35/56, 63%). Other seizure localisations were temporal lobe not otherwise specified (11/56, 20%), temporal lobe involving Heschl’s gyrus (4/56, 7%), frontal lobe (3/56, 5%), tempoparietal region (1/56, 2%), temporal and occipital lobes (1/56, 2%), and hemispheric onset (1/56, 2%). Involvement of Heschl’s gyrus was presumed if music provoked seizures (three patients) or if the patient heard music during the seizure (one patient).
Patients evaluated for seizure management were often medically refractory
Seizures were medically refractory in the majority (42/57, 74%). However, medically refractory epilepsy was significantly more frequent among patients with epilepsy in isolation who were evaluated for seizure management (34/39, 87%), compared with patients with epilepsy as part of an overlap syndrome who may have presented for management of SPSD or cerebellar ataxia rather than epilepsy (8/18, 44%) (p=0.0007).
Epilepsy surgery uniformly revealed gliosis and did not usually result in seizure freedom
The most common neuroimaging finding prompting consideration of epilepsy surgery was mesial temporal sclerosis, or MTS (9/62, 15%). Eight of 62 patients (13%) underwent epilepsy surgery (unilateral anterior temporal lobectomy, 7 patients; unilateral anterolateral temporal/frontal lobe resections, 1 patient). Neuropathological data was available for 5/8 patients, all of whom had gliosis reported. Two of 5 had pathological evidence of chronic inflammation noted (mild leptomeningeal, focal superficial cortical and perivascular chronic inflammation, 1 patient; ‘patchy chronic inflammation’ as per Mayo Clinic physician interpretation of outside neuropathology report, 1 patient). No more likely alternative aetiology for seizures (eg, malformation of cortical development) was reported in any patient. At last follow-up after surgery, only 2/8 (25%) obtained seizure freedom (one patient had focal seizures with preserved awareness up to 9 months after surgery that ceased with intravenous IG over two further years of follow-up, 1 patient had focal seizures with impaired awareness up to ten years after surgery that ceased with addition of clobazam over two further years of follow-up). The remaining six patients continued to have disabling seizures (ie, seizures limiting daily activities, requiring acute medical evaluation and/or leading to injury) after surgery.
Epilepsy was typically young-onset and chronic in disease duration
On stratification by core disease manifestation (table 1), median age at symptom onset ranged from 24 years (range: 5–56 years) in epilepsy to 59 years (range: 14–83 years) in cerebellar ataxia (p<0.0001). We examined the age at symptom onset of individual core disease manifestations in patients with overlap syndromes, and similarly found that the median age ranged from 33 years (range: 11–60 years) for epilepsy onset to 53 years for both cerebellar ataxia onset (range: 26–69 years) and SPSD onset (range: 19–70 years) (p<0.0001). The median total symptom duration recorded ranged from 42 months (range: 3–171 months) in cerebellar ataxia to 137 months (range: 3–552 months) in epilepsy (p<0.0001).
Epilepsy showed a trend toward less cerebrospinal fluid inflammation
On review of laboratory results (table 1), median serum and cerebrospinal fluid (CSF) anti-GAD65 titre did not differ significantly across core manifestations of GAD65 neurological autoimmunity. Patients with epilepsy had the lowest median CSF anti-GAD65 titre (2.5 nmol/L) and the lowest frequency of elevated CSF IgG index (0/22, 0%) among core disease manifestations, but these differences did not reach statistical significance (p=0.10 and p=0.17, respectively).
Limbic encephalitis
Patients with epilepsy uncommonly had neuroimaging evidence of LE
On MRI, medial temporal lobe T2-hyperintensity compatible with LE was seen in 10/62 (16%). These patients were classified separately as having LE and all had subacute-onset seizures/cognitive impairment. Only 1/10 (10%) were assessed at the Mayo Clinic within 3 months of disease onset, and 3/10 (30%) were assessed greater than 1 year after disease onset for management of sequelae of LE (ie, persistent seizures, cognitive difficulties).
Secondary manifestations of GAD65 neurological autoimmunity include cognitive impairment, myelopathy and brainstem dysfunction
Of the secondary disease manifestations cognitive impairment was the most common, being reported in 38/212 (18%). The predominant cognitive sphere impacted was short-term memory in 29/38 (76%), followed by working memory/attention in 6/38 (16%), and verbal fluency/expressive language in 3/38 (8%). Myelopathy was reported in 23/212 (11%), and manifested as upper motor neuron (UMN) signs (brisk tendon reflexes, extensor plantar responses and/or spasticity) in 19/23 (83%), followed by pyramidal weakness in 14/23 (61%) and bowel/bladder dysfunction in 4/23 (17%). No patient had spinal cord T2-hyperintensity or gadolinium enhancement indicative of myelitis. Concern for brainstem dysfunction was reported in 22/212 (10%) and was on the basis of oculomotor findings in all, including vertical misalignment (11/22, 50%), horizontal misalignment (7/22, 32%) and conjugate ophthalmoparesis (4/22, 18%). No patient had brainstem T2-hyperintensity or gadolinium enhancement indicative of brainstem encephalitis.
Secondary manifestations of GAD65 neurological autoimmunity clustered with specific core manifestations
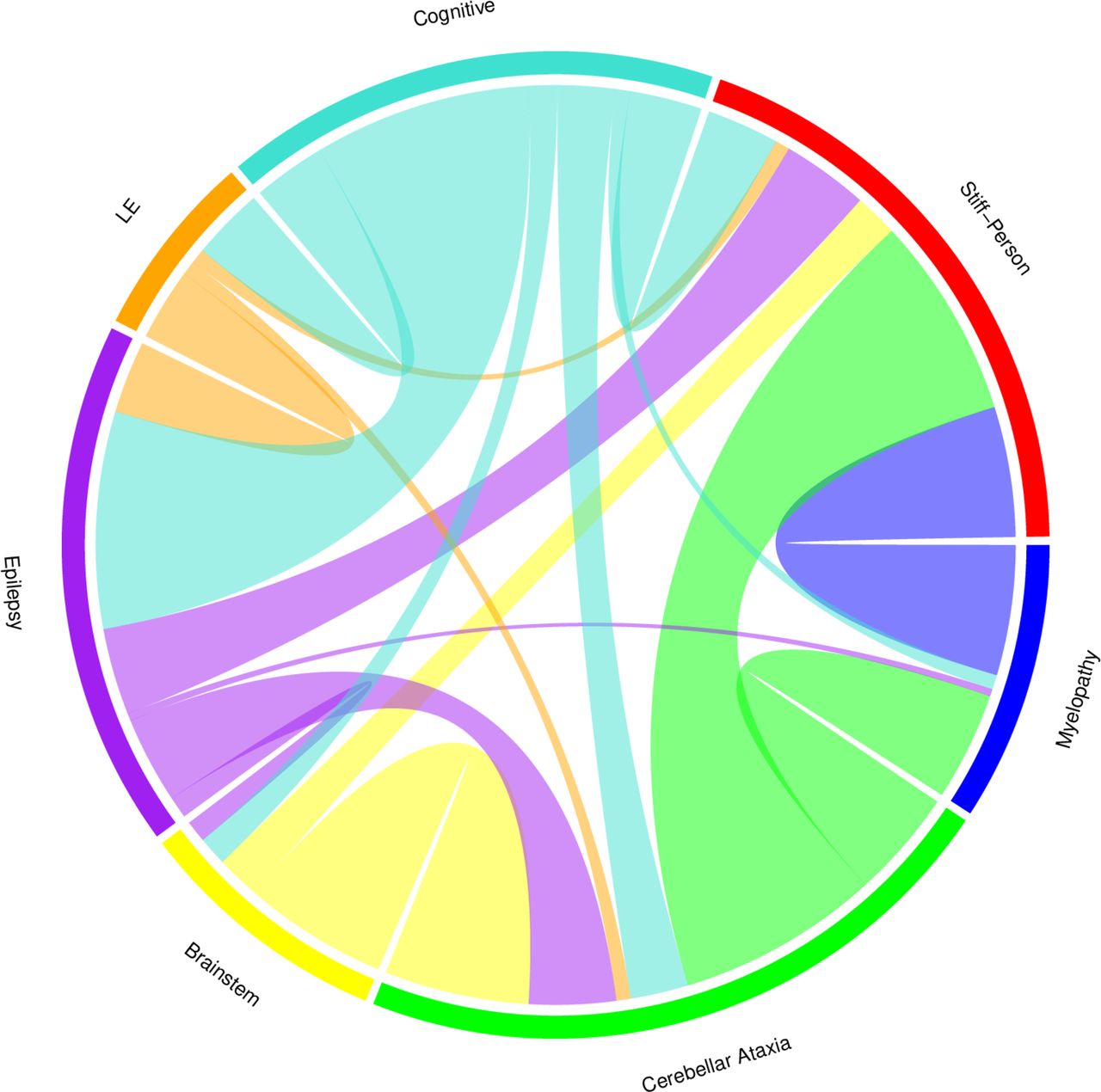
Secondary disease manifestations clustered with specific core disease manifestations: cognitive impairment with epilepsy/LE (N=30/38, 79%), myelopathy with SPSD (N=18/23, 78%), and brainstem dysfunction with cerebellar ataxia (N=20/22, 91%). The relationships among core and secondary disease manifestations are depicted via chord diagram (figure 2).

{kind=link}
{kind=link}
CHORD diagram depicting relationships among manifestations of GAD65 neurological autoimmunity in this chord diagram, Arcs representing the relationships among core manifestations (stiff-person spectrum disorder (SPSD), cerebellar ataxia, epilepsy and limbic encephalitis (LE)) and secondary manifestations (myelopathy, brainstem dysfunction and cognitive impairment) of GAD65 neurological autoimmunity are shown. The size of the Arc is proportional to the significance of the relationship. Orientation of disease manifestations around the CHORD diagram has been chosen to highlight significant overlap of neighbouring categories: cognitive impairment and epilepsy/LE, myelopathy and SPSD, and brainstem dysfunction and cerebellar ataxia. GAD65, glutamic acid decarboxylase-65.
An atypical presentation of GAD65 neurological autoimmunity: hyperkinetic movement disorders
Three patients had high-titre GAD65 antibodies but atypical presentations for GAD65 neurological autoimmunity (table 2). All had unilateral hyperkinetic movement disorders (dystonia, 2 patients; chorea, 1 patient). In patients with dystonia the onset was insidious, while in the patient with chorea onset was subacute. One patient with right lower extremity dystonia received intravenous IG and reported 90% improvement that was confirmed by the treating physician; however, dystonia recurred seven to 8 weeks after intravenous IG was discontinued due to intolerability.
Responses to immunotherapy and outcomes in GAD65 neurological autoimmunity
Responses to immunotherapy were stratified by core disease manifestation and are presented in table 3. Immunotherapy usage (corticosteroids, intravenous IG, PLEX, rituximab and cyclophosphamide) was not significantly different except for corticosteroid usage (p<0.0001). This was driven by infrequent corticosteroid usage in SPSD (7/44, 16%), the majority of whom received intravenous IG (38/44, 86%).
Responses to immunotherapy among 142 patients with GAD65 neurological autoimmunity
Patients with epilepsy received immunotherapy later and were least immunotherapy responsive
The median time from symptom onset to first immunotherapy ranged from 5 months (range: 1–22 months) in LE to 50.5 months (range: 1–324 months) in epilepsy (p<0.0001). The number of patients with sustained response to immunotherapy ranged from 5/20 (25%) in epilepsy to 32/44 (73%) in SPSD (p=0.002).
Complete response to immunotherapy was rare
Among all patients treated with immunotherapy, a complete response was reported in only 2/142 (1%); one patient had mild ataxic dysarthria that resolved after corticosteroids, and one patient had new-onset seizures with cortical-subcortical lesions on MRI that resolved after corticosteroids, intravenous IG and PLEX. In retrospect this patient’s clinicoradiographic presentation was concerning for co-existing for gamma-aminobutyric acid type A receptor encephalitis,26 but confirmatory testing for this antibody was not performed.
Presence of cerebellar ataxia and serum GAD65 antibody titre >500 nmol/L predicted poor outcome
Among patients with GAD65 neurological autoimmunity the mRS at last follow-up was as follows: 0, 2/212 (1%); 1, 28/212 (13%); 2, 61/212 (29%); 3, 65/212 (31%); 4, 49/212 (23%); 5, 3/212 (1%); 6, 4/212 (2%). Logistic regression analysis revealed that mRS >2 at first Mayo Clinic evaluation, cerebellar ataxia and serum GAD65 antibody titre >500 nmol/L were independent predictors of poor outcome (mRS >2) at last clinical follow-up (table 4).
Logistic regression analysis assessing predictors of poor outcome (MRS >2) at last clinical follow-up in 212 patients with GAD65 neurological autoimmunity
Discussion
This study of patients with GAD65 neurological autoimmunity provides numerous important insights into the disease. Through systematic review of all Mayo Clinic patients with high-titre GAD65 antibodies identified in our Neuroimmunology Laboratory over a 15-year period, we found that SPSD, cerebellar ataxia, epilepsy, and LE were core disease manifestations. Phenotypically, SPSD was usually classical in presentation, in keeping with previous studies.7 Cerebellar ataxia most often impacted gait, although limb and speech ataxia were also commonly reported. Among those with epilepsy, seizures typically originated from the temporal lobe. Interestingly, three patients had musicogenic epilepsy, suggesting patients with this rare form of reflex epilepsy should be considered for GAD65 antibody testing.27 28 With regard to immunotherapy-responsiveness, this differed significantly across core disease manifestations; SPSD was the most likely to respond to immunotherapy, while epilepsy was least immunotherapy responsive. We also determined that serum GAD65 antibody titre >500 nmol/L as well as cerebellar ataxia independently predicted poor outcome. An mRS >2 at first Mayo Clinic evaluation also independently predicted poor outcome, although the external validity of this finding to other centres requires further study.
Across core disease manifestations the age of symptom onset was youngest for epilepsy, indicating that a prior epilepsy diagnosis in a patient presenting with features of SPSD or cerebellar ataxia may be a clue to GAD65 neurological autoimmunity. Medically-refractory epilepsy was reported in the majority but was significantly more frequent among those with epilepsy in isolation who were evaluated for seizure management, compared with those with epilepsy as part of an overlap syndrome who may have been evaluated for management of SPSD or cerebellar ataxia. This suggests that referral bias may skew toward more severe epilepsy in publications, a finding that should be part of a balanced prognostic discussion in newly-diagnosed GAD65 epilepsy patients. LE was least-represented in our cohort, which likely reflects the rarity of this presentation as well as the primarily outpatient tertiary care setting of this study; patients with severe presentations of LE might have been less likely to travel to our facility. A cancer diagnosed within 5 years of symptom onset, which is the timeframe within which an associated neurological disorder is typically considered paraneoplastic,29 only occurred in 9/212 (4%). However, this differed significantly across core disease manifestations, with the highest rates of cancer in patients with cerebellar ataxia (6/55, 11%) and LE (1/7, 14%) as noted previously.30
Cognitive impairment, brainstem dysfunction and myelopathy were frequent accompaniments of GAD65 neurological autoimmunity but did not occur in isolation, hence their designation as secondary disease manifestations. This finding emphasises that patients with high-titre GAD65 antibodies who only have cognitive impairment, myelopathy or brainstem dysfunction should be thoroughly evaluated for alternative etiologies, because such presentations in isolation are not typical of GAD65 neurological autoimmunity. Secondary disease manifestations clustered intuitively with core disease manifestations: cognitive impairment with epilepsy/LE, myelopathy with SPSD, and brainstem dysfunction with cerebellar ataxia. Cognitive impairment was typically amnestic in keeping with medial temporal lobe dysfunction, as would be expected given the high rate of co-occurrence with temporal lobe epilepsy and LE.31 32 Myelopathic findings were most often reported in SPSD and usually manifested as UMN findings (brisk reflexes, extensor plantar responses, mild UMN pattern of weakness), in keeping with previous reports.7 The frequent coexistence of brainstem dysfunction with cerebellar ataxia on the basis of oculomotor findings could reflect more diffuse posterior fossa inflammation (‘rhombencephalitis’) in some patients, as well as the difficulties parsing out whether such findings are brainstem or cerebellar in clinical practice.33 34 Three patients had hyperkinetic movement disorders, suggesting this phenotype may be part of the spectrum of GAD65 neurological autoimmunity.35 However, given the paucity of cases, thorough evaluation for other causes of a hyperkinetic movement disorder in a patient with high-titre GAD65 antibodies is recommended.
On review of immunotherapy usage across core disease manifestations, only corticosteroid usage differed significantly. This was driven by the low usage of corticosteroids in patients with SPSD who instead largely received intravenous IG, which is likely due to randomised-controlled trial evidence for intravenous IG in SPSD.36 When evaluating immunotherapy-responsiveness, due to the retrospective nature of this study we were not able to implement standardised measures of disease severity when monitoring responses to immunotherapy. We thus chose to classify patients as having no response, partial response, near-complete response, or complete response to immunotherapy, based on the Mayo Clinic treating physician’s documentation. There is an element of subjectivity to this approach, but it has immediate translatability to clinical practice (eg, sustained response to immunotherapy is least often seen in epilepsy, response to immunotherapy is rarely complete) and is thus of clear utility to practitioners.37 Rates of sustained response to immunotherapy ranged from 73% in SPSD to only 25% in epilepsy, highlighting the recalcitrance of this disease manifestation.12 While the poor response to immunotherapy in patients with epilepsy and high-titre GAD65 antibodies may lead one to question whether or not anti-GAD65 is directly relevant to epilepsy aetiology, the high prevalence of epilepsy among these patients along with previously published series support a true disease association. Median time from symptom onset to first immunotherapy was longest for epilepsy (50.5 months), which may contribute to lack of immunotherapy-responsiveness. This delay to immunotherapy likely reflects epilepsy chronicity in GAD65 neurological autoimmunity, which in combination with the younger age of onset would explain the long median symptom duration recorded for epilepsy (137 months). The indolence of GAD65 epilepsy is unique compared with other autoimmune epilepsies, which usually present more rapidly.38 Inflammation was reported neuropathologically in only 2/5 patients with GAD65 epilepsy who underwent epilepsy surgery, and there was also a trend toward lower median CSF anti-GAD65 titre and less frequent elevated CSF IgG index among these patients compared with other core disease manifestations. Taken together, these findings may reflect a lack of inflammation in patients with chronic GAD65 epilepsy at the time they undergo clinical evaluation; whether a more prominent inflammatory response is present early on that may be more amenable to immunotherapy remains undetermined.
With regard to patient outcomes we found that serum GAD65 antibody titre >500 nmol/L and cerebellar ataxia were independent predictors of poor outcome (mRS >2). The mRS was chosen as a measure of disease outcome given its frequent usage in scoring neurological disability and relative ease of determination, but may skew towards poor outcomes among patients with disease manifestations that prominently affect gait (ie, cerebellar ataxia). Despite this limitation of the mRS, its broad applicability means that predictors of a poor outcome as defined by mRS >2 are helpful when discussing disease prognosis.
There are several limitations to this retrospective study. Clinical reporting of GAD65 antibodies in the Mayo Clinic Neuroimmunology Laboratory is based only on RIA, and so confirmation of high-titre GAD65 antibodies by a second assay (eg, rodent brain tissue indirect immunofluorescence, or TIIF) was not required for study inclusion. However, reporting of anti-GAD65 by TIIF is not routinely done, and so our approach is representative of clinical practice. Additionally, even serum positivity for anti-GAD65 by TIIF may occur in patients without GAD65 neurological autoimmunity,19 highlighting the challenge in determining what test methodology or cut-off best defines a clinically relevant high-titre GAD65 antibody result. Implementation of other test methodologies such as ELISA, immunoblot or cell-based assay to detect high-titre GAD65 antibodies in some laboratories has created the need for assay comparison studies, which is an area of active investigation in our laboratory. Based on our findings and that of the previous literature, high-titre GAD65 antibodies in serum are best viewed as necessary, but not sufficient for a diagnosis of GAD65 neurological autoimmunity.39
The presence of anti-GAD65 in CSF supports an autoimmune aetiology in the appropriate clinical context,39 which in keeping with our finding of anti-GAD65 CSF positivity in all patients who were classified as having GAD65 neurological autoimmunity. Calculation of intrathecal anti-GAD65 synthesis has recently been suggested as the most definitive laboratory evidence of GAD65 neurological autoimmunity.39 This calculation (which requires paired serum and CSF as well as albumin measurement to determine synthesis rate) is not performed in our testing laboratory, and was not required for study inclusion. While its calculation may aid in the determination of GAD65 neurological autoimmunity, it is not yet in widespread use and so systematic evaluation of its diagnostic utility in clinical practice is required. Given the lack of a diagnostic gold standard for GAD65 neurological autoimmunity that is independent of GAD65 antibody testing,39 rigorous clinical evaluation to rule out alternative diagnoses in patients with atypical features remains prudent.
Prior to study analysis, we excluded one-third of patients with high-titre GAD65 antibodies who were classified as not having GAD65 neurological autoimmunity due to non-neurological presentations (eg, isolated T1DM) or more likely alternative neurological diagnoses. This seemingly high number of excluded patients could in part reflect referral bias at our specialised tertiary care centre, which may be enriched with patients who have atypical presentations for GAD65 neurological autoimmunity and are ultimately determined to have more likely alternative diagnoses. Additionally, it is possible that some patients who were considered to have a more likely alternative diagnosis for their neurological presentation may have had contributory GAD65 neurological autoimmunity (eg, SPSD potentially contributing to stiffness/spasms in a patient with myotonia congenita, GAD65 cerebellar dysfunction potentially contributing to episodic vestibular symptoms in patients diagnosed as having more common vestibular disorders such as vestibular neuritis or migraine, or GAD65 epilepsy potentially contributing to seizure aetiology in a patient with febrile seizures who developed MTS). However, rigorous efforts to only include patients with the disease of interest in studies such as this is critical to prevent ‘phenotype creep’, whereby neurological features of alternative diagnoses are mistakenly assumed to broaden the clinical spectrum of a neural antibody based solely on seropositivity by an imperfectly specific assay.40 Our finding that high-titre GAD65 antibodies in serum are suggestive of, but not pathognomonic for GAD65 neurological autoimmunity emphasises the importance of clinical-serological correlation when enrolling patients in future studies of this disease.
Data availability statement
Data are available on reasonable request. Deidentified participant data will be made available to any qualified investigator on reasonable request directed to the corresponding author (NLZ).
Ethics statements
Ethics approval
This study was approved by the institutional review board of the Mayo Clinic, Rochester, Minnesota.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AB designed/conceptualised the study, acquired/analysed the data, drafted the manuscript and composed the tables/figures. ES, EPF, DD, AZ, SSS, AG, EN and AM acquired/analysed the data, and revised the manuscript for intellectual content. SJP and NLZ designed/conceptualised the study, acquired/analyzed the data, revised the manuscript for intellectual content and supervised the study.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests AB has no disclosures to report. ES has no disclosures to report. EPF is a site principal investigator in a randomised placebo-controlled clinical trial of Inebilizumab (A CD19 inhibitor) in neuromyelitis optica spectrum disorders funded by MedImmune/Viela Bio. He receives no personal compensation and just receives reimbursement for the research activities related to the trial. DD has a patent pending for Kelch-like protein 11 as a marker of neurological autoimmunity and has received research support from Grifols, Translational Research Innovation and Test Development Office and, Center for Clinical and Translational Science. DD has consulted for UCB and Astellas. All compensation for consulting activities is paid directly to Mayo Clinic. AZ has a patent pending for PDE10A-IgG as a biomarker of neurological autoimmunity. SS has no disclosures to report. AG has a patent pending for MAP1B IgG as a biomarker of neurological autoimmunity and small-cell lung cancer. EN has no disclosures to report. AM reports grants from Alexion, grants from Grifols, grants from Euroimmun, outside the submitted work; in addition, AM has a patent for Septin-5-IgG pending, a patent for PDE10A-IgG pending, a patent for MAP1B-IgG pending, and a patent for GFAP-IgG pending. SJP reports grants, personal fees and non-financial support from Alexion Pharmaceuticals; grants from Grifols, Autoimmune Encephalitis Alliance; grants, personal fees, non-financial support and other from MedImmune; SJP has a patent (patent #8889102) (application#12-678350) on neuromyelitis optica autoantibodies as a marker for neoplasia, and also a patent (patent #9891219B2) (application#12-573942) on methods for treating neuromyelitis optica (NMO) by administration of eculizumab to an individual that is aquaporin-4 (AQP4)-IgG autoantibody positive; SJP also has patents pending for the following IgGs as biomarkers of autoimmune neurological disorders (septin-5, Kelch-like protein 11, GFAP, PDE10A and MAP1B). NLZ has no disclosures to report.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.