Article Text

Abstract
Objective We aimed to investigate the influence of environmental risk factors for multiple sclerosis (MS) in different genetic contexts, and study if interactions between environmental factors and human leucocyte antigen (HLA) genes differ in magnitude according to heterozygocity and homozygocity for HLA-DRB1*15:01.
Methods Using population-based case–control studies (6985 cases, 6569 controls), subjects with different genotypes and smoking, EBNA-1 status and adolescent Body Mass status, were compared regarding MS risk, by calculating OR with 95% CI employing logistic regression. The interaction between different genotypes and each environmental factor was evaluated on the additive scale.
Results The effect of each DRB1*15:01 allele on MS risk was additive on the log-odds scale for each additional allele. Interaction between DRB1*15:01 and each assessed environmental factor was of similar magnitude regardless of the number of DRB1*15:01 alleles, although ORs were affected. When any of the environmental factors were present in DRB1*15:01 carriers without the protective A*02:01 allele, a three-way interaction occurred and rendered high ORs, especially among DRB1*15:01 homozygotes (OR 20.0, 95% CI 13.1 to 30.5 among smokers, OR 21.9, 95% CI 15.0 to 31.8 among those with elevated EBNA-1 antibody levels, and OR 44.3, 95% CI 13.5 to 145 among those who reported adolescent overweight/obesity).
Conclusions The strikingly increased MS risk among DRB*15:01 homozygotes exposed to any of the environmental factors is a further argument in favour of these factors acting on immune-related mechanisms. The data further reinforce the importance of preventive measures, in particular for those with a genetic susceptibility to MS.
Data availability statement
Data are available upon reasonable request. Anonymised data will be shared by request from any qualified investigator who wants to analyse questions that are related to the published article.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system (CNS) that arises from an interplay between genes and lifestyle/environmental factors. Gene variants with the strongest associations with MS risk are located within the human leucocyte antigen (HLA) complex.1 The main risk allele is DRB1*15:01, but several other alleles from other HLA regions influence the risk of MS independently of DRB1*15:01 status. A large number of gene loci outside the HLA complex have been associated with MS risk in large-scale genome-wide association studies, but each of these gene loci only make weak to modest individual contributions to disease susceptibility.2–4 So far, mapped gene variants explain nearly 50% of the heritability.4 Gene–environment interactions are likely to contribute to the so-called missing heritability in MS. Smoking increases MS risk by 50%, the combination of the genetic risk factors DRB1*15:01 and absence of A*02:01 increase MS risk fivefold, whereas the combination of smoking with both these genetic risk factors increases MS risk 13-fold (OR 12.7, 95% CI 10.8 to 14.9).5 Similar interactions have been demonstrated between the same MS associated HLA alleles, and both elevated EBNA-1 antibody levels and adolescent obesity.6–8 Since the effect of the DRB1*15:01 allele on the susceptibility to MS has been reported to be additive on the log-odds scale for each additional allele,2 we aimed to investigate the risks associated with the above-mentioned environmental factors in different genetic loads, and explore the interactive effects between DRB1*15:01 and the environmental factors in more detail by taking into consideration the number of DRB1*15:01 alleles a person has.
Methods
Study design and data collection
The present report is based on data from Epidemiological Investigation of Multiple Sclerosis (EIMS) and Genes and Environment in Multiple Sclerosis (GEMS), which are Swedish population-based case–control studies.
The study base comprised the Swedish general population aged 16–70 years.
EIMS recruited incident cases of MS from hospital-based and privately run neurology units. Cases were diagnosed by a neurologist located at the unit in which the case was entered. Two controls per case were randomly selected from the national population register, frequency matched for the case’s age in 5 year age strata, sex and residential area. If a control declined to participate or was not traceable, another control was selected using the same principles. The study period was April 2005 to June 2015.
GEMS identified prevalent cases from the Swedish National MS-registry.9 One control per case, matched by age, gender and residential area at the time of disease onset, was randomly selected from the national population register. The study participants were recruited between November 2009 and November 2011. All cases in both studies were diagnosed according to the McDonald criteria.10 11
All participants in both studies were asked to provide blood samples and those who did not donate blood were excluded in the present report. The number of study subjects in each study is presented in online supplemental eTable 1. Ethical approvals for EIMS and GEMS were obtained from the Regional Ethical Review Board at Karolinska Institute. All participants gave their informed consent to participate in the studies.
Supplemental material
Data collection and exposure information
Information regarding environmental exposures and lifestyle factors was collected using standardised questionnaires. The response rate was 93% for cases and 73% for controls in EIMS, and 82% for cases and 66% for controls in GEMS.
Information on smoking was obtained by asking about current and previous smoking habits. The year of disease onset among cases was defined as the index year. The controls were given the same index year as their corresponding case. Smoking habits were only considered before and at the index year. Subjects were classified as ever smokers if they had smoked before or during the index year, and as never smokers if they had never smoked before or during the index year.
Information was obtained regarding current height and body weight at age 20 years. Using current height, we calculated adolescent body mass index (BMI) by dividing weight in kilograms by height in metres squared. The WHO’s definitions of overweight and obesity were used. A subject with a BMI equal to or more than 25 was considered overweight and a person with a BMI of 30 or more was considered obese.
Genotyping and measurement of EBNA-1 antibody levels
HLA-DRB1 and HLA-A alleles were determined using the MS replication chip4 which is based on an Illumina exome chip to which approximately 90 000 custom markers were added with extra high density in the HLA region. Classical four-digit HLA alleles were imputed with HLA*IMP:02.12
For participants included before August 2013, a multiplex serological assay was used for detection of IgG antibodies against the EBNA-1 peptide segment (aa 385–420),13 which has been identified as the main EBNA-1 fragment associated with MS risk.14 Dual-laser flow-based detection was used to quantify the antibodies as units of median flourescence intensity. EBNA-1 antibody levels were dichotomised into high and low EBNA-1 antibody levels based on the median flourescence intensity among controls.
Statistical analysis
Subjects with different genotypes and smoking habits were compared with regard to MS risk, by calculating OR with 95% CI using logistic regression models. In the same manner, OR of MS were calculated for subjects with different genotypes and EBNA-1 status, and adolescent BMI status, respectively. DRB1*15:01 was entered as a categorical variable.
We investigated the interaction between DRB1*15:01 and each of the environmental factors with regard to MS risk, among DRB1*15:01 heterozygotes and homozygotes, respectively. Additive interaction was defined as departure from additivity of effects and evaluated by calculating the attributable proportion due to interaction (AP) together with 95% CI, using the delta method.15 16 We also calculated the product terms for the interactions between HLA-DRB1*15:01 and each environmental factor in logistic regression models to assess interaction on the multiplicative scale.
All analyses were adjusted for study, age, sex, residential area, ancestry and when appropriate for A*02:01 (as a categorial variable), smoking (ever or never), EBNA-1 antibody status (low or high EBNA-1 antibody levels, or unknown) and adolescent BMI (normal weight, overweight, obese or unknown). Ancestry was dichotomised into Nordic or non-Nordic origin. We also adjusted the analyses for the following MS-associated HLA alleles; DRB1*03:01, DRB1*13:03, DRB1*08:01, B*44:02, B38:01, B44:02, DQA1*01:01, DQB1*03:02 and DQBI*03:01.2 Homozygote correction was made for DRB1*03:01 and when appropriate for A*02:01. Furthermore, adjustment was made for two SNPs from within the extended major histocompatability complex (MHC) region from 29.9 to 33.6 Mb on chromosome 6; rs9277565[T] and rs2229092[C] which have previously been associated with MS.2
The analyses were further adjusted for a large number of potential confounding variables that were not kept in the final analyses since they only had minor influence on the results. These variables were educational level (no postsecondary education, postsecondary education without university degree, or university degree), passive smoking (ever or never), snuff use (ever or never), alcohol consumption and ultraviolet radiation (UVR) exposure. Alcohol consumption was categorised into subgroups based on the amount of alcohol intake per week: low consumption (<50 g/week for women and <100 g/week for men), moderate consumption (50–112 g/week for women and 100–168 g/week for men) and high consumption (>112 g/week for women and >168 g/week for men). We used the same cutoffs as those used by Statistics Sweden,17 a government agency that produces official statistics.
We constructed a continuous variable for sun exposure, based on three questions regarding exposure to UVR where each answer alternative was given a number ranging from 1 (the lowest exposure) to 4 (the highest exposure). The numbers were added together and we thus acquired a value between 3 and 12.18
As a sensitivity analysis, we used principal component analysis (PCA) vectors based on 3736 ancestrally informative markers to adjust for population stratification. In this analysis, population outliers were excluded (85 cases and 66 controls). All analyses were conducted using Statistical Analysis System V.9.4.
Results
The mean age at onset was 34.4 years in EIMS and 32.1 years in GEMS and the median duration from the initial appearance of symptoms indicative of MS to study enrolment was 4.5 years in EIMS and 18.0 years in GEMS.
The HLA-DRB1*15:01 allele
The increased risk of MS conveyed by the DRB1*15:01 allele was additive on the log-odds scale for each additional allele. Compared with DRB1*15:01 negative subjects, the adjusted OR of developing MS was 3.7 (95% CI 3.3 to 4.1) for DRB1*15:01 heterozygotes and 7.8 (95% CI 6.4 to 9.5) for DRB1*15:01 homozygotes (table 1).
OR with 95% CI of developing MS among subjects categorised by DRB1*15:01 status, compared with DRB1*15:01 negative subjects
Interaction between DRB1*15:01 and each environmental factor, with adjustment for A*02:01
Smoking
The OR of MS associated with smoking among DRB1*15:01 negative subjects was 1.5 (95% CI 1.4 to 1.7). The combination of DRB1*15:01 and smoking increased the risk of MS approximately sixfold among DRB1*15:01 heterozygotes and 12-fold among DRB1*15:01 homozygotes with ORs displaying non-overlapping CIs, compared with never smokers without the DRB1*15:01 allele (table 2, online supplemental eTable 2).
OR and AP with 95% CI of developing MS among subjects with different numbers of DRB1*15:01 alleles exposed to the assessed lifestyle/environmental factor, compared with non-exposed DRB1*15:01 negative subjects
EBNA-1 antibody levels
High EBNA-1 antibody levels among DRB1*15:01 negative subjects was associated with an increased risk of MS (OR 2.6, 95% CI 2.3 to 2.9), compared with DRB1*15:01 negative subjects with low EBNA-1 antibody levels. DRB1*15:01 in combination with elevated EBNA-1 antibody levels increased the risk of MS 10-fold among DRB1*15:01 heterozygotes and 20-fold among DRB1*15:01 homozygotes, compared with DRB1*15:01 negative subjects with low EBNA-1 antibody levels (table 2, online supplemental eTable 3). The CIs were non-overlapping.
Adolescent BMI
Adolescent overweight/obesity was associated with an OR of 1.4 (95% CI 1.2 to 1.6) among DRB1*15:01 negative subjects. Compared with DRB1*15:01 negative subjects with adolescent BMI below 25 kg/m2, the risk of MS among those who reported adolescent overweight/obesity was increased fivefold among DRB1*15:01 heterozygotes, and 12-fold among DRB1*15:01 homozygotes (table 2, online supplemental eTable 4). The CIs were non-overlapping.
An interaction on the additative scale was observed between DRB1*15:01 and each of the environmental factors regarding risk of developing MS. Interestingly, the magnitude of the attributable proportion was similar regardless of the number of DRB1*15:01 alleles. Interactions on the multiplicative scale were non-significant (table 2, online supplemental eTables 2–4).
Interaction between DRB1*15:01, absence of A*02:01 and each environmental factor
As stated before, the increased risk of MS conveyed by the DRB1*15:01 allele was additive on the log-odds scale for each additional allele. An interaction occurred between DRB1*15:01 and absence of A*02:01 that was similar among DRB1*15:01 heterozygotes (AP 0.26, 95% CI 0.17 to 0.35) and DRB1*15:01 homozygotes (AP 0.24, CI 0.003 to 0.5) (table 3).
OR with 95% CI of developing MS among subjects categorised by DRB1*15:01 and A*02:01 status
When any of the environmental factors were present, a three-way interaction occurred among the triple exposed that rendered high ORs, especially among DRB1*15:01 homozygotes (table 4 and online supplemental eTables 5–7.
OR and AP with 95% CI of developing MS among A*02:01 negative subjects with different DRB1*15:01 status exposed to the assessed lifestyle/environmental factor, compared with non-exposed A*02:01 positive and DRB1*15:01 negative subjects
The results presented in online supplemental eTables 5–7 are illustrated in figure 1.
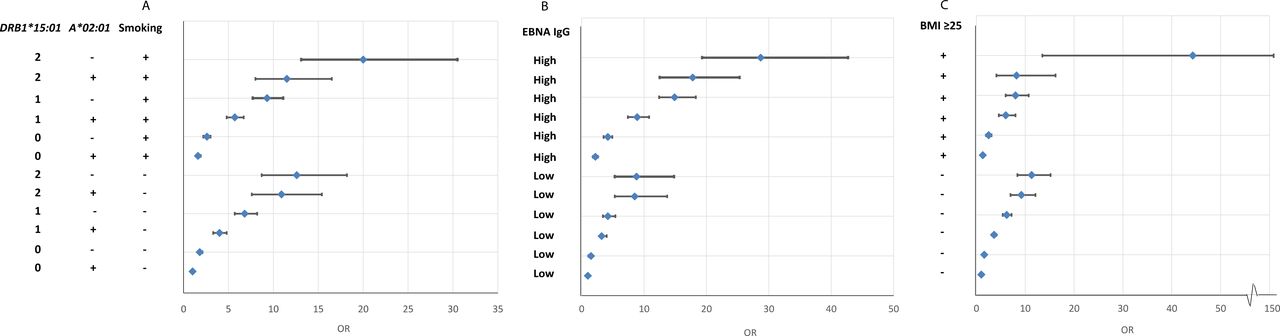
{kind=link}
OR of developing MS for subjects with different combinations of DRB1*15:01, A*02:01 and environmental exposures (smoking, EBNA-1 status and adolescent BMI, respectively), compared with unexposed subjects without the genetic risk factors. Based on data from online supplemental eTables 5–7. BMI, body mass index; MS, multiple sclerosis.
Our results remained almost identical after excluding population outliers and adjusting the analyses for PCA vectors.
Discussion
We demonstrate that synergistic effects between DRB1*15:01 on one side, and smoking, elevated EBNA-1 antibody levels and adolescent overweight/obesity on the other side take place with similar magnitudes among DRB1*15:01 heterozygotes and homozygotes. However, in the presence of any of the environmental factors, drastically increased ORs were evident among DRB*15:01 homozygotes compared with heterozygotes.
The effect of each DRB1*15:01 allele on the susceptibility to MS is additive on the log-odds scale, and may reflect that disease development is facilitated by surface expression of the antigen presenting molecules encoded by this allele.19 An increased surface expression of MS risk class II molecules may thus have a major effect on the ability of cells to present so far undetermined peptide antigens activating CD4+ T cells relevant for MS pathogenesis.
HLA molecules are also involved in regulating thymic selection of the mature T-cell repertoire. By central tolerance mechanisms, potentially autoaggressive T cells with a propensity to recognise CNS autoantigens may to a higher degree survive in DRB1*15:01 positive subjects.
The mechanism for the protective association of A*02:01 carriage is unclear. The molecule encoded by this allele presents antigen to CD8+ T cells. One might consider the presentation of autoantigens resulting in anti-inflammatory cytokines which has been demonstrated in animal models of MS.20 21 Hypothetically, other class I alleles may activate disease driving CD8+ T cells. In addition, expression of A*02:01 molecules may increase negative selection of CNS autoreactive T cells or modulate their autoreactivity.22 Absence of A*02:01 may thus increase the risk of autoreactive T cells inducing an immune response against the self-antigen.
The biological explanations for the interactions between MS-associated HLA genes and the assessed environmental factors are far from clear. Smoking initiates chronic inflammation in the lungs and induces post-translational modifications of peptides that increase their likelihood of activating autoimmune CD4+ T cells.23 24 Altered peptides in the lungs with structural similarities to CNS antigens could promote a CNS-directed autoimmune response in genetically predisposed individuals. The lungs also hold potentially autoaggressive effector and memory T cells that may become activated by smoke-induced inflammation and enter the CNS after assuming migratory properties.25 An interaction on the additive scale has repeatedly been observed between the same MS risk HLA genes and several lung-irritating agents regarding risk of developing MS,26 27 which supports the view that lung-irritation and inflammation are critically involved in the pathways to MS among smokers. Epigenetic modifications induced by smoking may also contribute to mediate the interaction between smoking and HLA alleles in MS development.28
The DRB1*15:01 allele may also influence the risk of MS by compromising the immune response to Epstein-Barr virus (EBV).29 It has been suggested that infected B cells migrate to a particular target organ, depending on the preferential peptide binding by allelic variants of MHC class II molecules.30 By cross-reactivity, structural similarity between EBV and CNS antigens may induce an autoaggressive T-cell response and subsequently lead to MS in genetically susceptible individuals.8 31–33 The study of EBV–HLA interaction is further complicated by the difference in EBV reactivity between HLA-DRB1*15:01 carriers and non-carriers.14
In case of the synergistic effect between HLA genes and obesity, we primarily consider a chronic, low-grade inflammation induced by the secretion of inflammatory mediators driven by adipose tissue macrophages.34 The Th1 promoting effects of obesity may increase the risk of an HLA restricted activation of autoaggressive CD4+ T cells that target CNS autoantigens.
From a public health perspective, the impact of environmental factors on MS risk is considerable and preventive measures are essential. Approximately 20% of all MS cases in Sweden have been estimated to be attributable to smoke exposure, whereas this number increased to more than 40% in DRB1*15 positive and A*02 negative cases.35 The prevalence of childhood overweight and obesity has increased during the last decades36 and may, at least to some extent, explain the increasing incidence of MS. Accordingly, prevention of smoking and childhood obesity may contribute to reduce MS risk. Several clinical trials have evaluated the effect of different vaccine strategies against EBV. While some of them lowered the rate of infectious mononucleosis, none of them protected from infection.37 It is also uncertain whether vaccination will lower the viral load among those who become infected with EBV. Further research is needed to better understand the immune mechanisms that are critical to prevent EBV infection.
Our studies were designed as case–control studies and information on exposures and lifestyle factors was collected retrospectively. EIMS recruited newly diagnosed cases in order to minimise recall bias. The potential for recall bias is higher in GEMS which used prevalent cases, but the magnitude of memory errors does probably not differ between cases and controls. Both the EIMS and GEMS questionnaires covered a wide range of questions regarding many environmental exposures and no part of the questionnaires was given prime focus.
A potential selection bias may arise during the recruitment of cases and controls. Since the public healthcare system in Sweden provides equal free of charge access to medical services for all citizens, almost all cases of MS are referred to neurological units. In both studies, the risk of selection bias was minimised by the population-based design. Although the participation rate was lower among controls, selection bias is probably modest because the prevalence of life style factors, such as smoking and alcohol consumption, among the controls was consistent with that of the general population.16 Furthermore, there were no significant differences with respect to age, gender or smoking habits between those who donated blood and those who did not. The interaction between environmental exposures and risk genes also alleviates some potential biases in the interpretation of the influence from these factors on MS risk, since HLA alleles are unlikely to determine exposure habits. We thus believe that our findings are not affected by bias to a large extent.
In conclusion, the strikingly increased MS risk among DRB*15:01 homozygotes exposed to any of the environmental factors is a further argument in favour of these factors acting on immune-related mechanisms. The data further reinforce the importance of preventive measures, in particular for those with a genetic susceptibility to MS.
Data availability statement
Data are available upon reasonable request. Anonymised data will be shared by request from any qualified investigator who wants to analyse questions that are related to the published article.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TO and LA contributed equally.
Contributors Conception and design of the study: AKH, JH, TO and LA. Acquisition of data: AKH, LA, JH, NB, JB, TW and PS. Statistical analysis: AKH. Drafting a significant portion of the manuscript or figures: AKH, TO, LA and IK. All authors commented on the draft and approved the final version of the manuscript.
Funding The study was supported by grants from the Swedish Research Council (2016-02349); from the Swedish Research Council for Health, Working Life and Welfare (2015-00195 and 2019-00697) and the Swedish Brain Foundation (FO2020-0077).
Competing interests Hedström has nothing to disclose. Hillert received honoraria for serving on advisory boards for Biogen and Novartis and speaker’s fees from Biogen, Merck-Serono, Bayer-Schering, Teva and Sanofi-Aventis. He has served as principal investigator for projects sponsored by, or received unrestricted research support from, Biogen, Merck-Serono, TEVA, Novartis and Bayer-Schering. Waterboer, Brenner, Butt and Strid have nothing to disclose. Olsson received honoraria for advisory boards an unrestricted MS research grants from Biogen, Novartis, Sanofi, Roche and Merck. Alfredsson reports grants from Swedish Research Council, grants from Swedish Research Council for Health Working Life and Welfare, grants from Swedish Brain Foundation, during the conduct of the study; personal fees from Teva, personal fees from Biogene Idec, outside the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.