Article Text

Abstract
The objective of this paper is to evaluate available evidence for each step in autoimmune encephalitis management and provide expert opinion when evidence is lacking. The paper approaches autoimmune encephalitis as a broad category rather than focusing on individual antibody syndromes. Core authors from the Autoimmune Encephalitis Alliance Clinicians Network reviewed literature and developed the first draft. Where evidence was lacking or controversial, an electronic survey was distributed to all members to solicit individual responses. Sixty-eight members from 17 countries answered the survey. Corticosteroids alone or combined with other agents (intravenous IG or plasmapheresis) were selected as a first-line therapy by 84% of responders for patients with a general presentation, 74% for patients presenting with faciobrachial dystonic seizures, 63% for NMDAR-IgG encephalitis and 48.5% for classical paraneoplastic encephalitis. Half the responders indicated they would add a second-line agent only if there was no response to more than one first-line agent, 32% indicated adding a second-line agent if there was no response to one first-line agent, while only 15% indicated using a second-line agent in all patients. As for the preferred second-line agent, 80% of responders chose rituximab while only 10% chose cyclophosphamide in a clinical scenario with unknown antibodies. Detailed survey results are presented in the manuscript and a summary of the diagnostic and therapeutic recommendations is presented at the conclusion.
- autoimmune encephalitis
- paraneoplastic syndrome
- neuroimmunology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Autoimmune encephalitis (AE) comprises a group of non-infectious immune-mediated inflammatory disorders of the brain parenchyma often involving the cortical or deep grey matter with or without involvement of the white matter, meninges or the spinal cord.1–4 The original description of AE was based on paraneoplastic conditions related to antibodies against intracellular onconeuronal antigens such asANNA-1/anti-Hu.5 6 These ‘classical’ antibodies are non-pathogenic but represent markers of T-cell-mediated immunity against the neoplasm with secondary response against the nervous system. In recent years, an increasing number of antibodies targeting neuronal surface or synaptic antigens have been recognised such as N-MethylD-Aspartate Receptor (NMDAR)-antibody and Leucine-richglioma inactivated (LGI1)-antibody.1 Most of these surface antibodies have been shown to be likely pathogenic and are thought to mediate more immunotherapy-responsive forms of AE and have less association with tumours. Specific types of encephalitis can occur in the setting of antibodies against oligodendrocytes (eg, anti-myelin oligodendrocyte glycoprotein (MOG) brainstem encephalitis) or astrocytes (eg, anti-aquaporin-4 (AQP4) diencephalic encephalitis, anti-glial fibrillary acidic protein (GFAP) meningoencephalitis). In addition, some AE patients do not have any identifiable antibodies (seronegative) representing a disease category with novel, yet to be identified antibodies or T-cell mediated disease. Online supplemental appendix S1 contains a list of the commercially available neuronal autoantibodies (NAAs).
Supplemental material
Recent epidemiological studies suggest that AE is possibly as common as infectious encephalitis with an estimated prevalence rate of 13.7/100 000.7 The rapidly advancing knowledge of new antibodies and their associated syndromes has created a new and growing field of autoimmune neurology.8 However, advances from the laboratory bench have not been paralleled by advancement in clinical practice, leading to a large knowledge gap and many unanswered questions regarding the acute and long-term management of AE. The heterogeneity of AE presentation and findings on ancillary testing hinder large-scale clinical trials and limit the quality of evidence behind AE management.
The objective of this paper is to evaluate available evidence for each step in AE management and provide expert opinion when evidence is lacking. Although the turnaround time of commercial NAAs panels may improve in the near future, currently these results are often unavailable at the time of early evaluation and management. Moreover, current commercial NAAs panels are inherently limited in their ability to diagnose AE, given the ever-growing numbers of antibodies identified and the likelihood of T-cell mediated pathogenesis in some cases. Consequently, clinicians have to approach AE initially as a clinical entity when deciding on investigations and treatment.1 Long-term management can then be modified according to the type of antibody identified, if any. Therefore, the aim of this paper is to emphasise the practical acute and long-term management of AE as a broad category rather than focusing on individual antibody syndromes. Another important goal is to represent the practice of experienced clinicians from different clinical and geographical backgrounds.
Methods
Core authors from the Autoimmune Encephalitis Alliance Clinicians Network (AEACN) developed the first draft of this paper (HA, JCP, SI, RCD, EPF, PG, AJ, YL, AR-G, IR, SJP and MJT). The AEACN is comprised of self-identified clinicians with interest and clinical expertise in AE management listed by the AE Alliance, a non-profit organisation founded by AE patients and families to establish a supportive community for patients and caregivers, enhance clinical collaboration, and facilitate AE scientific research. The AEACN includes a multidisciplinary international group of adult and paediatric neurologists, rheumatologists, psychiatrists, neuropsychologists and other specialists with real-life experience in AE management. The authors of the first draft reviewed available literature to compile existing evidence for every step in AE management. Where evidence was lacking or controversial, an electronic survey was distributed to all AEACN members to solicit individual responses. The survey questions were strategically planned to look at initial treatment, continued care and finally long-term management. After adding survey results to the manuscript, the updated version was circulated to all participating AEACN members for edits and further suggestions.
Survey results
The survey was distributed to 147 Clinical members. Sixty-eight (46%) members responded including the core authors. The most represented specialty/subspecialty of the respondents was neuroimmunology (66%), followed by general neurology (21%), paediatric neurology (16%), epilepsy (9%), behavioural/cognitive neurology (6%), hospital neurology-neurohospitalist (6%), neuromuscular neurology (6%), paediatric rheumatology (6%), neurocritical care (4%), psychiatry (4%), movement disorders (3%), general paediatrics (3%) and one specialist (1.5%) each of the following: autonomic disorders, adult rheumatology and paediatric critical care. Twenty-five members (37%) indicated more than one subspecialty.
Clinicians from 17 countries participated including USA (69%), Brazil (4%), Canada (3%), China (3%), Spain (3%), Argentina, Australia, Indonesia, Israel, Italy, the Netherlands, the Philippines, Singapore, South Korea, Switzerland, Turkey and the UK (countries listed in a descending order based on the number of responders and alphabetically when the number of responders was equal). Of the total participating members, 88% practiced at academic tertiary referral centres and 76% were active in AE clinical research or scholarly publications. The participating members indicated personal clinical experience with an average of 7.3 AE subtypes (range 1–13 subtypes, median 8 subtypes). In total, 9% of the participating members were affiliated with reference neuroimmunology laboratories with NAAs testing capabilities. The results of individual survey questions are presented under the corresponding sections of AE management. The final draft was approved by all participating AEACN members after four rounds of revisions. The paper aimed to answer prespecified clinical questions as detailed below.
Data availability statement
The results of the survey are partially summarised in figure 1 and the detailed responses of all survey questions are published as online supplemental document 2.
Supplemental material

AEACN survey results for acute and bridging therapy. AE, autoimmune encephalitis; AEACN, Autoimmune Encephalitis Alliance Clinicians Network; IVMP, intravenous methylprednisolone; IVIg, intravenous Ig; PLEX, plasma exchange.
Section 1: diagnosis of AE
When to suspect AE clinically?
A detailed history and examination is the first and most important step in AE diagnosis. The immune reaction in AE often results in acute or subacute presentation of a duration less than 3 months.1 Chronic presentations are only seen in some of these conditions, especially LGI1, Contactin-associatedprotein-like 2 (CASPR2), Dipeptidyl-peptidase-likeprotein 6 (DPPX) and Glutamicacid decarboxylase 65 (GAD65)-antibody encephalitis, and should otherwise raise suspicion of a neurodegenerative disease or other etiologies.9 Likewise, hyperacute presentations are also atypical and a vascular aetiology should be considered in those cases. A recurrent course may point towards an autoimmune aetiology but unlike the typical relapsing-remitting course of multiple sclerosis and systemic inflammatory disorders, AE relapses are rare and often result from insufficient treatment or rapid interruption of immunotherapy. A monophasic course is more common in idiopathic AE while a progressive course may be seen in some paraneoplastic syndromes especially paraneoplastic cerebellar degeneration, which tends to plateau after the cancer is treated. Patients with known cancer or those at increased cancer risk (smokers, the elderly, and patients with rapid unintentional weight loss) are prone to paraneoplastic AE, while patients with personal or family history of other autoimmune disorders are at increased risk of idiopathic AE.10 A preceding viral infection, fever or viral-like prodrome is common.11 AE may be triggered by herpes simplex virus (HSV) encephalitis or certain immune-modulating therapies such as TNFα inhibitors, and immune-checkpoint inhibitors (ICIs)—the latter can cause an accelerated form of paraneoplastic encephalitis in patients with advanced cancer.1 12 Table 1 shows practical classification concepts in AE.
Proposed classification concepts in autoimmune encephalitis
The immune reaction in AE is usually diffuse, resulting in multifocal brain inflammation and occasionally additional involvement of the meninges, spinal cord and/or the peripheral nervous system.3 6 This diffuse inflammation may or may not be detectable on ancillary testing but it usually results in a polysyndromic presentation which is a clinical hallmark of AE. Although some antibodies have been linked to stereotypical symptoms (eg, oromandibular dyskinesia, cognitive/behavioural changes, and speech and autonomic dysfunction in NMDAR-antibody encephalitis, faciobrachial dystonic seizures in LGI1-antibody encephalitis, etc), there is significant symptom overlap between all antibodies and all forms of AE.1 11 Symptoms vary according to the anatomical localisation of inflammation and there are several clinical-anatomical syndrome categories in AE as summarised in table 2.
Anatomical-clinical syndromes of autoimmune encephalitis
What investigations should be ordered when AE is suspected?
After AE is suspected clinically, a detailed workup is needed to confirm the diagnosis and exclude competing possibilities like infective encephalitis or systemic/metabolic causes. In most cases, the workup starts with brain imaging and cerebrospinal fluid (CSF) analysis. The diagnostic algorithm follows the structure summarised in figure 2 and detailed below:
Aim 1: confirming the presence of focal or multifocal brain abnormality suggestive of encephalitis
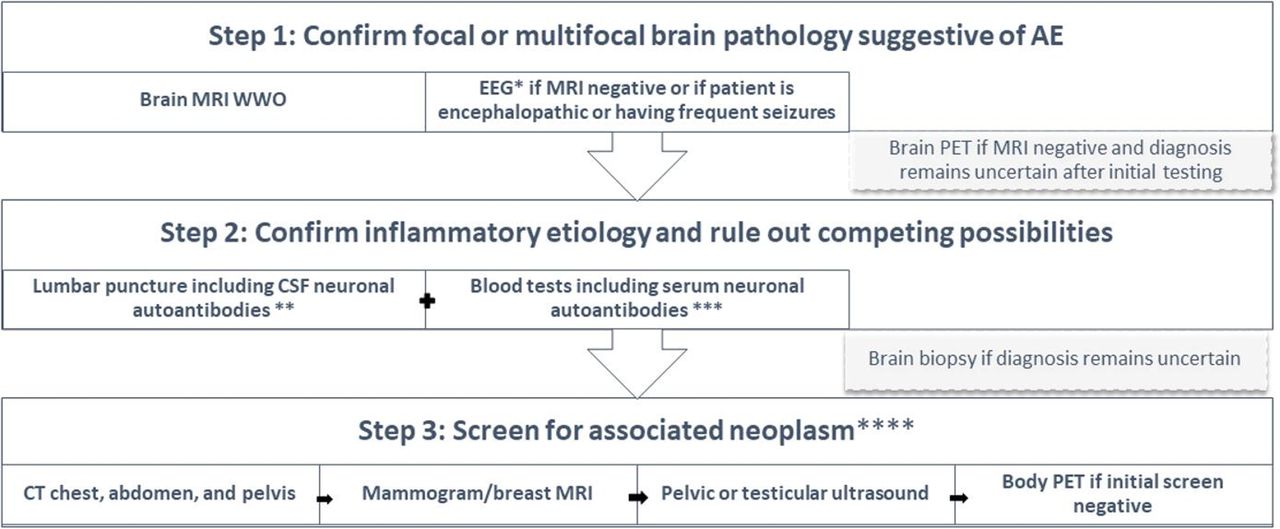
Diagnostic algorithm for autoimmune encephalitis. *EEG can confirm focal or multifocal brain abnormality and rule out subclinical seizures. **In addition to neuronal autoantibodies, cerebrospinal fluid should be tested for infections, inflammatory markers (IgG index and oligoclonal bands), and in some cases cytology. ***In addition to neuronal autoantibodies, the differential diagnosis generated based on MRI results will guide what blood tests to send. ****In most cases, general neoplasm screening starts with CT then other screening modalities are added until a neoplasm is found or eventually ruled out. If the clinical picture is highly suggestive of a specific neoplasm, a targeted screening approach could be implemented (eg, starting with pelvic ultrasound if the clinical picture is suggestive of anti-NMDAR encephalitis). AE, autoimmune encephalitis; EEG, electroencephalogram; MRI WWO, MRI with or without contrast; PET, positron emission tomography.
Brain MRI
In addition to ruling out alternative diagnoses, standard Brain MRI with contrast can show changes consistent with one or more of the AE anatomical syndromes (table 1 and figure 3). According to the 2016 AE clinical criteria by Graus et al, the presence of bilateral limbic encephalitis is the only MRI finding sufficient to diagnose definite AE in the correct clinical setting (eg, negative CSF viral studies) even in absence of NAAs.1 All other MRI patterns (cortical/subcortical, striatal, diencephalic, brainstem, encephalomyelitis and meningoencephalitis) can support possible or probable AE unless the NAAs panel is positive for a clinically relevant antibody.1 2 Diffuse or patchy contrast enhancement suggestive of inflammation is seen in a few patients while intense enhancing lesions are unlikely in AE.3 9 Rare findings include focal or extensive demyelination, meningeal enhancement, and rarely cortical diffusion restriction (often related to secondary seizures). Brain MRI may also be normal. Patients with initially negative MRI may show changes suggestive of AE on repeat MRI a few days later. Gadolinium should be avoided during pregnancy. Table 3 shows the main differential diagnoses for each of the AE anatomical syndromes.
Differential diagnosis of autoimmune encephalitis anatomical syndromes and suggested additional testing

Anatomical subtypes of autoimmune encephalitis. (A) Limbic encephalitis, (B) cortical/subcortical encephalitis, (C) striatal encephalitis, (D) diencephalic encephalitis, (E) brainstem encephalitis (arrow), (F) meningoencephalitis (arrow).
Importantly, brain MRI can also help exclude alternative diagnoses such as acute stroke, neoplasm or Creutzfeldt-Jacob disease (CJD), although AE MRI changes can sometimes mimic some of these entities. Unilateral, and to a lesser extent bilateral, inflammation of the mesial and non-mesial temporal lobe as well as the orbitofrontal cortex on FLAIR or DWI sequences supports herpetic encephalitis over AE.13 Parenchymal haemorrhage on gradient echo sequence is more common in herpetic encephalitis than AE although this difference did not reach statistical significance in one underpowered study comparing the two types of encephalitis.14
In some related immune-mediated conditions, the diagnosis can be inferred from typical MRI patterns such as radial perivascular enhancement in autoimmune GFAP astrocytopathy and punctate brainstem/cerebellar enhancement in chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS).15 16
Electroencephalogram
Electroencephalogram (EEG) is commonly performed in patients with suspected AE to exclude subclinical status epilepticus in encephalopathic patients or to monitor treatment response in patients with seizures. AE is a major cause of new onset refractory status epilepticus (NORSE), which can be convulsive or non-convulsive.17 EEG can also provide evidence of focal or multifocal brain abnormality when MRI is negative which would support encephalitis over metabolic encephalopathy.1 Findings suggestive of AE include focal slowing/seizures, lateralised periodic discharges and/or extreme delta brush, which is occasionally seen in NMDAR-antibody encephalitis.18 Frequent subclinical seizures are commonly identified in LGI1-antibody encephalitis but patients may also have a normal EEG including those with classical faciobrachial dystonic seizures (FBDS).19 20 Although a normal EEG does not exclude AE, it can support primary psychiatric disorders when investigating patients with isolated new psychiatric symptoms. EEG can also help differentiate AE from CJD.
Brain fluorodeoxyglucose positron emission tomography
In the event of a negative brain MRI and clinical uncertainty despite high suspicion of AE, obtaining a brain fluorodeoxyglucose positron emission tomography (FDG-PET) can confirm focal or multifocal brain abnormality in the correct clinical setting.21 It can also substitute for MRI when MRI is contraindicated. In case series, brain FDG-PET was more sensitive than MRI and may reveal brain abnormalities at an earlier stage of the disease.22 Bilateral temporal hypermetabolism (in seropositive or seronegative limbic encephalitis) and bilateral occipitoparietal hypometabolism (in NMDAR-antibody encephalitis) are among the most common patterns seen and may prove useful biomarkers for specific syndromes. Importantly, further studies are needed to better differentiate AE metabolic patterns from neurodegenerative and neuroinfectious syndromes. In addition, immunosuppressants, anaesthetics and antiseizure therapies, commonly administered to AE patients, can also alter cortical metabolism. Seizures can also cause hypermetabolic changes on FDG-PET. The lack of specificity and the limited availability of FDG-PET are barriers against the wide utilisation of this technique in AE diagnosis.
Aim 2: confirming an autoimmune inflammatory etiology and excluding other possibilities
Following assessment for focal or multifocal brain abnormality by MRI or other studies, additional investigations are indicated to confirm AE and exclude other possibilities. Testing can be guided by the clinical-anatomical syndrome to narrow down the scope of investigations as shown in table 3.
CSF analysis
This is the most important test in AE evaluation and is usually the second step in the workup after brain MRI. Regardless of MRI findings, all patients with suspected encephalitis require a lumbar puncture (LP) unless there is a significant contraindication (eg, risk of herniation on brain imaging). In some cases, inflammatory CSF may be the only abnormality found on initial testing serving as the sole indication for empiric immunotherapy after infection is excluded. If timely brain MRI is not possible due to patient agitation or lack of access, clinicians should proceed with LP after a screening head CT so as not to delay immunotherapy. CSF analysis should include cell count and differential, protein, glucose, CSF/serum glucose ratio, albumin quotient, IgG index and synthesis rate, oligoclonal bands, broad viral studies including HSV1/2 PCR and varicella zoster virus (VZV) PCR and IgG/IgM, bacterial/fungal cultures when appropriate, cytology, flow cytometry, NAAs panel (eg, Autoimmune encephalopathy/encephalitis panel, etc), and in some cases, prion disorder panel (preferably RTQuIC when available). Common CSF findings in AE include mild to moderate lymphocytic pleocytosis (commonly 20–200 cells but can be as high as 900 cells with some antibodies), hyperproteinorrachia, and in some cases, elevated IgG index and/or IgG synthesis rate and positive intrathecal oligoclonal bands (unmatched in the serum).1 23 These findings in the setting of negative infectious and cytological studies support an immune-mediated aetiology but would not differentiate AE from other immune-mediated conditions (eg, neurosarcoidosis) so clinical correlation is always needed. In many patients, testing NAAs in both CSF and serum is needed because CSF detection is more sensitive for some antibodies (eg, NMDAR and GFAP antibodies) while serum is more sensitive for other antibodies (eg, onconeuronal, LGI1, and AQP4 antibodies).1 If the clinical picture is highly suggestive of an antibody with a higher serum sensitivity (eg, FBDS suggestive of LGI1-antibody encephalitis), then it might be reasonable to avoid CSF testing in clinical situations where CSF sampling is challenging. Although symptomatology can guide which neuronal antibodies (or antibody panels) to test for in some patients, it may be most practical to send the most comprehensive panel especially in patients with less defined presentations. This is because there is a significant syndromic overlap between most of these antibodies and because more than one antibody can coexist in the same patient.24 Notably, routine CSF studies may be normal in some AE patients and this does not exclude the diagnosis when other parameters are consistent with AE; therefore, testing NAAs panels is recommended in case of high clinical suspicion even if the CSF is normal.25
Blood tests
In addition to testing NAAs in the serum, several blood tests are often needed to exclude other competing etiologies. Test selection can be guided by the MRI anatomical pattern as shown in table 3 but some tests may be useful in case of negative MRI such as antithyroid antibodies, toxicology screen, ammonia, vitamin B1/B12 levels, HIV, inflammatory markers, antinuclear antibodies, extractable nuclear antigen antibodies, antiphospholipid and lupus anticoagulant antibodies, immunoglobulins and metabolic and hormonal panels when appropriate.1 Monitoring sodium level is important since hyponatraemia is common with certain AE subtypes such as LGI-1 antibody encephalitis.19 Blood samples should be collected prior to treatment with intravenous immunoglobulins or plasmapheresis to avoid false positive or false negative results.
Brain biopsy
Most AE cases with normal brain MRI or typical MRI patterns (limbic, striatal, etc) do not require a brain biopsy. Rarely, a brain biopsy may be needed for atypical or mass-like lesions to exclude neoplastic or other possibilities especially when all other investigations point away from autoimmunity.1 Pathological findings in AE are nonspecific and include T-cell and/or B-cell perivascular and parenchymal infiltrates along with secondary gliosis.26
Aim 3: screening for an associated neoplasm
It is nearly impossible to predict whether AE is paraneoplastic or non-paraneoplastic based on symptoms as both AE subtypes present similarly. Therefore, cancer screening should be considered in most adult AE patients at time of presentation.24 If the patient has a known history of cancer typically associated with paraneoplastic syndromes then a paraneoplastic aetiology is presumed, and repeat cancer screen is indicated to identify recurrence or progression. In patients with cancer history not typically associated with paraneoplastic neurologic syndromes (eg, basal cell skin cancer, prostate cancer), repeat cancer screen may unmask a new different tumour. The most common neoplasms associated with AE include small cell lung cancer, thymic neoplasm, breast cancer, ovarian teratoma or carcinoma, testicular teratoma or seminoma, neuroblastoma and lymphoma.24 In some patients, the suspicion of associated neoplasm may be high based on certain demographic factors (eg, smoking history or advanced age) or typical clinical picture (NMDAR-antibody encephalitis associated with ovarian teratoma). Although some antibodies have stronger cancer association than others (eg, antibodies against intracellular antigens), the implicated antibody is usually unknown at the time of first presentation. The following screening modalities are available:
CT chest, abdomen and pelvis
Initial screening with CT of the chest, abdomen and pelvis with contrast is a reasonable approach given its lower cost compared with FDG-PET and since it provides more structural details of the neoplasm (if present) to guide biopsy and further surgical intervention if indicated. A major limitation of CT-based screening is its low sensitivity for early breast and testicular cancers.24 In addition, CT is not preferred in children and pregnant women; and pelvic CT is not preferred for women in childbearing age in general. Moreover, CT contrast dye may be contraindicated due to renal impairment or dye allergy. In these situations, additional or alternative means (eg, MRI) of cancer screening are required. It is to be noted, however, that CT iodine-based dye is relatively safer in pregnant women compared with MRI gadolinium-based dye.
Mammogram and breast MRI
Breast cancer is a common source of paraneoplastic syndromes in females, and a mammogram should be performed if the initial CT screen is negative.24 Patients with a strong family history of breast cancer and those who are not up to date with their regular mammograms are a special concern. If mammogram is negative but the suspicion of breast cancer is high, then breast MRI may improve sensitivity of cancer detection.
Pelvic or testicular ultrasound or MRI
Young and middle age adults with a typical clinical picture of NMDAR-antibody encephalitis should be specifically screened for teratoma by a transvaginal or transabdominal pelvic ultrasound (or testicular ultrasound in males).24 In female patients with ataxic presentation (suggestive of PCA1/Yo antibody), pelvic ultrasound can screen for ovarian carcinoma. Likewise, in males with ataxia and other brainstem symptoms (suggestive of Ma and Kelch-like Protein-11 Antibodies), testicular ultrasound may reveal the associated neoplasm.27 Pelvic MRI may be useful if ultrasound is equivocal. Extraovarian and extratesticular germ cell tumours may be detected on CT-based or MRI-based general cancer screening.
Whole body FDG-PET scan
Whole body FDG-PET can be more sensitive for early neoplasms when initial CT screen is negative or inconclusive and the suspicion of cancer is high (eg, smoker elderly patient, classic paraneoplastic presentation).24 It can also be used as the initial screening tool when there is a contraindication to high resolution CT or iodine contrast. Insurance coverage can be an obstacle and insurers should consider fewer restrictions on FDG-PET in AE patients given the high likelihood of a coexisting cancer in those patients.
Section 2: acute treatment
Intensive care unit needs
The main indications for intensive care unit (ICU) admission in AE include refractory status epilepticus, severe dysautonomia and respiratory compromise (eg, from brainstem involvement, associated neuromuscular syndrome or medication-induced hypoventilation).28 It is important for ICU clinicians to distinguish central non-infectious fevers caused by the primary disease from infectious processes. Careful monitoring and management of blood pressure and heart rate fluctuations is critical in patients with severe dysautonomia. A temporary pacemaker may be needed in patients with severe dysrhythmia until the dysautonomia improves. Patients with severe hyponatraemia may require controlled slow correction of sodium levels to avoid central pontine myelinolysis. In most cases, hyponatraemia is related to inappropriate antidiuretic hormone secretion and fluid restriction is sufficient. In rare occasions with massive inflammation and brain oedema, intracranial pressure monitoring and management may be indicated. AE patients are often subject to high doses of sedation, antiseizure medications, and other symptomatic therapies so monitoring for drug toxicity in the ICU is imperative.
Empiric antimicrobial treatment
In many encephalitis patients, differentiating infectious from autoimmune aetiologies may be difficult prior to CSF analysis and therefore starting empiric antimicrobials with CNS coverage is always recommended until infection is excluded. The common practice is to start CNS doses of intravenous acyclovir and standard coverage for bacterial meningitis. Antibiotics and acyclovir can later be discontinued if CSF bacterial and HSV/VZV studies return negative.
Acute immunotherapy
Several retrospective studies have shown that early and aggressive immunotherapy is associated with better outcomes in AE patients.1 29 The 2016 AE clinical criteria emphasise the importance of starting immunotherapy once AE is highly suspected and infectious etiologies are excluded based on CSF results (cell-count, glucose, viral PCR, gram stain). It is impractical and potentially hazardous to delay immunotherapy until AE is confirmed by a positive antibody. There are no robust clinical trials comparing the different modalities of acute immunotherapy; therefore, the choice of the initial therapy may be based on anecdotal evidence and factors related to the specific syndromic presentation and comorbidities as shown in figure 4 and detailed below:

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Therapeutic algorithm for autoimmune encephalitis. *Relative contraindications to steroids include uncontrolled hypertension, uncontrolled diabetes, acute peptic ulcer and severe behavioural symptoms that worsen with corticosteroid therapy. **Steroid-responsive conditions include faciobrachial dystonic seizures suggestive of LGI1-antibody encephalitis, autoimmune encephalitis in the setting of immune checkpoint inhibitors, central demyelination, autoimmune GFAP astrocytopathy, chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids, and steroid-responsive encephalopathy associated with autoimmune thyroiditis. ***High thromboembolic risk includes patients with known or suspected cancer, smoking history, hypertension, diabetes, hyperlipidaemia and hypercoagulable states. Ab, antibody; AE, autoimmune encephalitis; Ag, antigen; IVMP, intravenous methylprednisolone; IVIg, intravenous Ig; IL-6: interleukin 6; NORSE, new-onset refractory status epilepticus; PLEX, plasma exchange.
High-dose corticosteroids
Empiric treatment with intravenous methylprednisolone at a dose of 1 g per day for 3–7 days is a common reasonable approach to achieve initial immunosuppressive and anti-inflammatory effect in AE patients.1 It is also the preferred approach in presentations known to be specifically corticosteroid-responsive namely demyelinating pattern on MRI (suggestive of AE overlap with demyelinating syndromes),30 or dotted or radial enhancement (suggestive of CLIPPERS or autoimmune GFAP astrocytopathy, respectively).15 16 Patients with FBDS suggestive of LGI1-antibody encephalitis may also show a dramatic response to corticosteroids.19 Patients with known or highly suspected paraneoplastic AE associated with classical onconeuronal antibodies are thought to have a primarily T-cell mediated inflammation making corticosteroids, theoretically, a preferred option for immunosuppression over intravenous IG or plasma exchange (PLEX). However, paraneoplastic conditions associated with classical onconeuronal antibodies are often resistant to immunosuppression and tend to respond best to cancer therapy. A notable exception are patients who develop accelerated paraneoplastic AE in the setting of ICI treatment. These patients may be particularly responsive to corticosteroids given their inhibitory effect on T-cell overactivity which is the pathogenic hallmark of ICI-associated immune adverse events; however, second-line therapies may also be needed in some cases.12
On our AEACN survey, 84% of responders chose corticosteroids alone (65%) or in combination with other agents (19%) for initial immunotherapy in patients with a general AE presentation. Likewise, 74% of responders chose corticosteroids for initial immunotherapy for patients presenting with FBDS suggestive of LGI1-antibody encephalitis, alone (58%) or in combination with other agents (16%). For NMDAR-antibody encephalitis, corticosteroids remained the most popular choice on the survey. However, the percentage was lower selected only by 63% of responders either alone (35%) or combined with other agents (28%) indicating a larger diversity among specialists when selecting first-line therapy in those patients. Similar diversity was present for treatment of known or highly suspected paraneoplastic AE; whereas corticosteroids remained the most popular choice, it was chosen by only 48.5% of responders, alone (29%) or combined with other agents (19%) (see online supplemental document S2).
One theoretical disadvantage to corticosteroids in AE is their potential for causing initial worsening of behavioural/psychiatric symptoms hampering a timely evaluation of treatment response although in most cases, corticosteroids may actually improve these symptoms. The use of corticosteroids may also be difficult in patients with common comorbidities such as uncontrolled hypertension or diabetes. Some experts recommend avoiding corticosteroids in patients with known GAD65-antibody associated neurological syndromes for fear of inducing type-1 diabetes but this concern remains theoretical without confirmatory studies. In patients with atypical or mass-like lesions on brain MRI in whom primary CNS lymphoma is on the differential diagnosis, corticosteroids should be delayed so as not to interfere with pathology results if a biopsy is considered during hospitalisation. Similar precautions are advisable when systemic autoimmunity such as sarcoidosis is on the differential.
Intravenous Ig
Intravenous Ig (IVIg) at a dose of 2 g/kg over 2–5 days is a relatively easy-to-use and timely option for fast immunomodulation when corticosteroids are contraindicated or when the clinical picture is suggestive of or known to be related to antibody-mediated disease (eg, probable or definite NMDAR-antibody encephalitis).29 IVIg can be more readily available than PLEX in some centres and it does not require a central line. A recent randomised blinded study showed IVIg efficacy over placebo in controlling seizures in a small number of patients with LGI1-antibody and CASPR2-antibody AE.31 On our AEACN survey, IVIg was the most popular acute immunotherapy if corticosteroids are contraindicated chosen by 41% of responders. Also 40% of responders indicated choosing IVIg alone or in combination with corticosteroids and other immunotherapies for acute therapy if the clinical picture was suggestive of NMDAR-antibody encephalitis.
A downside to IVIg is its association with increased thromboembolic risk. Therefore, IVIg should be used with caution in patients with known or suspected paraneoplastic AE or other risk factors for thrombosis (eg, heavy smokers and the elderly). In addition, the aetiology of paraneoplastic AE associated with antibodies against intracellular antigens is thought to be cell-mediated rather than antibody-mediated rendering the use of IVIg in this setting potentially ineffective. On our survey only 25% of responders indicated using IVIg in known or suspected paraneoplastic AE. The use of IVIg may also worsen coexisting hyponatraemia due to volume expansion, which may potentially predispose to brain oedema and worsening mental status.32
Plasma exchange
PLEX (5–10 sessions every other day) is an effective option for acute immunomodulation when corticosteroids are contraindicated or ineffective. In a small retrospective study, patients with NMDAR-antibody encephalitis treated with both corticosteroids and PLEX had better improvement in the modified Rankin score than those treated with corticosteroids alone,33 which is similar to the results in other antibody-mediated conditions like NMOSD.34 PLEX may be particularly effective in AE cases with associated central demyelination or coexisting NMOSD. It provides a potentially faster immunomodulation in patients with severe or fulminant presentations. It has no known psychiatric side effects and does not increase the risk of thromboembolism except for line-related thrombosis. Major limitations include increased bleeding risk, volume shifts (which can be problematic in dysautonomic patients), and the need for central line placement (in some institutions) with its associated risks. In addition, it is less suitable for agitated patients.
Combined first-line therapies
If the initial clinical picture is severe (eg, NMDAR-antibody encephalitis, NORSE, severe dysautonomia), clinicians may consider using combined first-line therapies from the beginning despite the lack of high quality evidence to support this practice. On our AEACN survey, combination therapy was the second most popular choice after corticosteroids alone if the clinical picture was suggestive of NMDAR-antibody encephalitis chosen by 28% of responders, and for unspecified AE (19%). More commonly, combination therapy is done sequentially if there is no meaningful response to the initial agent (eg, adding IVIg and/or PLEX after completing corticosteroids). On the survey, 62% of responders chose adding a different first-line therapy if the initial agent was ineffective while 26% chose going directly to a second-line agent. Other options like adding a second round or prolonging the duration of the same first-line agent were less popular.
Second line agents
If there is no meaningful clinical or radiological response to optimised first-line therapy after 2–4 weeks, the addition of a second-line agent with both rapid and sustained immunosuppressive effects can improve the outcome.29 However, the exact definition and timing of treatment responsiveness is not well defined and some clinicians may anecdotally choose earlier initiation of second-line agents. Both rituximab and cyclophosphamide have been used as second-line agents for rescue therapy in AE with good results.29 Rituximab is less toxic than cyclophosphamide and therefore is preferentially considered by most clinicians although it may not be as effective for cell-mediated inflammation as in the case of antibodies against intracellular antigens. However, although rituximab acts mainly on B-cells, it indirectly suppresses T-cell activity by reducing B-cell drive to T-cells. In most newly diagnosed cases, it is hard to determine clinically whether AE is antibody or cell-mediated before the antibody results are available. Some clues may help the clinician come to a preliminary hypothesis regarding aetiology (eg, FBDS or typical NMDAR-antibody encephalitis presentation suggest antibody-mediated AE while patients with known or increased cancer risk are more likely to have cell-mediated AE). Based on these clues, clinicians may decide to use rituximab or cyclophosphamide as a second-line agent if antibody results are delayed or if there is no access to antibody testing. Common rituximab dosing regimens include 375 mg/m2 weekly for 4 weeks or two doses of 1000 mg 2 weeks apart. Common dosing regimen of cyclophosphamide include 600–1000 mg/m2. A few case series have shown response to proteasome inhibitors that block plasma-cell generation (bortezomib), interleukin (IL)-6 inhibition (tocilizumab), or low dose IL-2 in patients who did not respond quickly to conventional second-line agents.35–37 However, the evidence behind these non-conventional rescue therapies remains limited and more research is needed to confirm their effectiveness in refractory AE. A clinical trial of ocrelizumab (a humanised anti-CD20 monoclonal antibody with a similar mechanism of action to rituximab) is currently recruiting, and a clinical trial of bortezomib is underway (www.clinicaltrials.gov, accessed 13 April 2020).
When a second-line agent is used in the acute setting, it also serves as a bridging therapy to prevent early relapses that might happen if immunosuppression is abruptly discontinued.38 Prognostication and clinical severity tools are being developed to help select patients who would benefit from conventional and non-conventional second-line agents such as the anti-NMDAR Encephalitis 1-year Functional Status score and the Clinical Assessment Scale in Autoimmune Encephalitis.39 40
On the AEACN survey, 50% of responders indicated they would consider adding a second-line agent in the acute setting only if there was no response to more than one first-line agent, 32% indicated adding a second-line agent if there was no response to one first-line agent, while only 15% indicated using a second-line agent in the acute setting on all patients regardless of the response to first-line therapy. As for the preferred second-line agent, 80% of responders chose rituximab while only 10% chose cyclophosphamide in a clinical scenario with unknown antibodies and no clinical clues for aetiology.
Conclusion
In this first part of the best practice recommendations, we covered the clinical presentation, diagnostic workup and acute management of AE guided by published studies and the results of the AEACN survey providing updated recommendations for management of patients with suspected AE. The second part will follow with a focus on bridging therapy, symptomatic treatment and maintenance immunotherapy. A discussion of the limitations will be presented at the end of the second part. A summary of the best practice recommendations for AE diagnosis and acute management is presented in box 1.
Best practice recommendations summary for acute management of autoimmune encephalitis (AE)
Evaluate the likelihood of AE relative to the patient’s clinical picture.
Perform brain MRI and/or EEG to look for focal or multifocal brain abnormality.
Perform lumbar puncture to support inflammatory aetiology and rule out infective/neoplastic causes. Test oligoclonal bands, IgG index, IgG synthesis rate and neuronal autoantibodies in the cerebrospinal fluid (CSF).
Send blood tests to rule out other potential causes guided by neuroanatomical and clinical data. Test neuronal autoantibodies in the serum.
Consider brain FDG-PET when there is a high clinical suspicion of AE and other paraclinical studies are uninformative.
Perform cancer screening with CT chest, abdomen, and pelvis with contrast in relevant cases (or MRI when CT is contraindicated or not preferred). If negative, consider further testing with mammogram/breast MRI, pelvic ultrasound, and/or whole body FDG-PET guided by the clinical presentation and each patient’s specific cancer risk factors.
Once infection is ruled out based on basic CSF results (eg, number of cells) and if biopsy for primary CNS lymphoma or neurosarcoidosis is not a consideration, start acute immunotherapy with high dose corticosteroids (or IVIG or PLEX if steroids are not preferred or contraindicated).
If there is no clinical, radiological or electrophysiological improvement by the end of the initial treatment cycle, add IVIG or PLEX. Consider IVIG first in agitated patients and in those with bleeding disorders. Consider PLEX first in patients with severe hyponatraemia, high thromboembolic (or cancer) risk, and if there is associated brain or spinal demyelination.
Consider starting with a combination therapy of steroids/IVIG or steroids/PLEX from the beginning (as opposed to sequentially) in patients with severe initial presentation (eg, severe NMDAR-antibody presentation, new onset refractory status epilepticus, severe dysautonomia, etc).
If there is no clinical or radiological improvement 2–4 weeks after completion of combined acute therapy, consider starting a second-line agent when the clinical suspicion is high and/or a clinically relevant antibody is present.
Consider rituximab in known or highly suspected antibody-mediated autoimmunity (eg, NMDAR-antibody encephalitis) and consider cyclophosphamide in known or highly suspected cell-mediated autoimmunity (eg, classical paraneoplastic syndrome).
If no clear objective or subjective evidence of improvement with conventional second-line therapies, consider novel approaches such as tocilizumab or bortezomib although there is only minimal evidence to support their use.
Start bridging therapy with gradual oral prednisone taper or monthly intravenous Ig or intravenous methylprednisolone. Avoid steroid taper or implement a shorter taper in vague cases with poor response to initial immunosuppressive therapy or when immunosuppression may impose higher risks than benefits (eg, patients with cancer or active infection).
Ethics statements
Acknowledgments
The authors would like to thank Kimberley de Haseth, Director of Programs at the Autoimmune Encephalitis Alliance for coordinating the communication between the AEACN members and for distributing the survey.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SJP and MJT are joint senior authors.
Twitter @ANG_Oxford, @Dr_GalenoRojas
Correction notice This article has been correcte since it appeared Online First. In the Contributors and Collaborators sections, author Jonathan Santoro has been updated to Jonathan D Santoro.
Collaborators The following members of the Autoimmune Encephalitis Alliance Clinician Network answered the survey and contributed to the manuscript: Rawan Tarawneh, Heather Van Mater, Eyal Muscal, Ilene Ruhoy, Yaacov Anziska, Erin Longbrake, Susa Benseler, Cynthia Wang, Michelle Apperson, Raffaele Iorio, Mateus Mistieri Simabukuro, Ning Zhong, Stephan Rüegg, Amanda Piquet, Jonathan Kuo, Bahadir Konuskan, Elena Frid, Joseph Deng, Wendy Mitchell, GenaLynne Mooneyham, Riwanti Estiasari, Yuhei Chiba, Melanie Alarcio, Velda Han, Jon P. Williams, Michael Sweeney, Tania Cellucci, Kyle Blackburn, Marisa Klein-Gitelman, Jonathan D Santoro, Raymond Suarez, Jose Irazuzta, Staley Brod, Ann Hyslop, Katrina Irene Manibog, Domingo Escudero, William Horace Noland, Stacey Clardy, Soe Mar, and William Kilgo.
Contributors HA conceptualised the project, designed the survey, wrote the first draft, handled submission and was responsible for the final approval of the manuscript. JCP conceptualised the project, cowrote the first draft and was responsible for the final approval of the manuscript. SI, SJP and MJT revised and cowrote the first group draft, revised the final manuscript for intellectual content and were responsible for the final approval of the manuscript. RCD and EPF revised and cowrote the first group draft and revised the final manuscript for intellectual content. PG, AJ, YL, AR-G and IR conceptualised the project and cowrote the first draft. BA, DRB, MB, PPC, MF-F, AG, EG, S-TL, MM, AMM, GR, SS, AV and SV revised the manuscript for intellectual content and cowrote the final draft.
Funding Open access funding was provided by the Autoimmune Encephalitis Alliance. The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests HA is a consultant for Roche/Genentech, which manufactures rituximab and tocilizumab that were discussed in this paper. HA is a consultant for Bristol-Myers Squibb, which manufactures cyclophosphamide that is discussed in this paper. SI is a coapplicant and receives royalties on patent application WO/210/046716 (UK patent No. PCT/GB2009/051441) licensed to Euroimmun for the development of assays for leucine-rich glioma inactivated protein 1 and other voltage-gated potassium channel complex antibodies discussed in this paper. AG has a patent for MAP1B autoantibodies as biomarkers of neurological autoimmunity and small cell lung cancer. S-TL is a consultant for GC Pharma, which manufactures IVIg that was discussed in this paper and for Advanced Neural Technologies which operates several neuronal autoantibody panels. SV receives research support from Quest Laboratories Diagnostics, which offers several commercial neuronal autoantibody panels, and from Genentech (rituximab) and Grifols (IVIG). SJP has a patent Patent# 8 889 102 (Application#12-678350) - Neuromyelitis Optica Autoantibodies as a Marker for Neoplasia issued, and a patent Patent# 9 891 219B2 (Application#12-573942) - SJP has a patent pending for GFAP, Septin-5, Kelch11 and MAP1B autoantibodies as biomarkers of neurological autoimmunity. Some of these antibodies are discussed in this paper. MJT has filed a patent for methods for typing neurologic disorders and cancer, and devices for use therein, and has received an unrestricted research grant from Euroimmun AG.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Editorial commentary
- Neuro-inflammation