Article Text

Abstract
Objectives Alemtuzumab is effective in patients with active multiple sclerosis but has a complex safety profile, including the development of secondary autoimmunity. Most of patients enrolled in randomised clinical trials with alemtuzumab were either treatment naïve or pretreated with injectable substances. Other previous disease-modifying treatments (DMTs) were not used in the study cohorts, and therefore, associated risks might yet remain unidentified.
Methods We retrospectively evaluated a prospective dual-centre alemtuzumab cohort of 170 patients. We examined the baseline characteristics as well as safety and effectiveness outcomes, including the time to first relapse, the time to 3 months confirmed disability worsening and the time to secondary autoimmunity.
Results The regression analysis showed that, among all previously used DMTs, the pretreatment with fingolimod (n=33 HRs for the time to first relapse (HR 5.420, 95% CI 2.520 to 11.660; p<0.001)) and for the time to worsening of disability (HR 7.676, 95% CI 2.870 to 20.534; p<0.001). Additionally, patients pretreated with fingolimod were more likely to experience spinal relapses (55% vs 10% among previously naïve patients; p<0.001) and had an increased risk of secondary autoimmunity (HR 5.875, 95% CI 2.126 to 16.27; p<0.001).
Conclusion In the real-world setting, we demonstrated suboptimal disease control and increased risk of secondary autoimmunity following alemtuzumab, among patients previously treated with fingolimod. These data can provide guidance for improving MS therapeutic management.
Data availability statement
Data are available on reasonable request. Anonymised data will be shared on reasonable request from qualified investigators.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Despite the approval of several disease-modifying treatments (DMTs) within the past decade, the therapeutic management of relapsing-remitting multiple sclerosis remains challenging. Most of patients are initially treated with low-risk first-line treatments and are switched to highly active therapies, which are potentially associated with more severe side effects, only if further disease activity occurs.
Alemtuzumab (ALEM) is an anti-CD52 monoclonal antibody,1 which was shown to be highly efficacious in controlling the disease activity, among both treatment naïve patients (CARE-MS I) and those, who had poor response to first line DMT (CARE-MS II).2 3 Patients enrolled in the CARE-MS II trial had been previously treated mainly with beta-interferon (IFN) or glatiramer acetate, although a minority had received natalizumab (NTZ), azathioprine or mitoxantrone.2 Ongoing real-world cohorts, such as the TREAT-MS registry,4 provide important information regarding the use of ALEM and demonstrated that its effectiveness proportionally reduces with the number of previously administered DMTs.5
Despite a variety of new DMTs having entered the clinical routine, real-world data on specific treatment sequences remains sparse and the optimisation of the escalating therapeutic management remains short of general consensus. The effectiveness and safety profile of ALEM, among patients pretreated with NTZ, has been suggested to be consistent to the core study results.6 7 However, evidence of the ALEM effectiveness, among patients who previously failed to respond to fingolimod (FTY) has been conflicting. While a British case series suggested that the escalation to ALEM might not achieve a good control of the disease activity,8 more retrospective analyses demonstrated good effectiveness and safety profile of ALEM following FTY pretreatment.9 10 In addition, data on the effect of ALEM among patients previously treated with teriflunomide, dimethyl fumarate (DMF) or ocrelizumab are currently missing.
Real-world data are also needed to assess the potential impact of DMT sequencing on the immune system in order to assist the decision making process in clinical practice. This is particularly relevant to the ALEM, as its use has been recently restricted by medical authorities to a subset of pretreated and highly active patients, because of its complex safety profile, including infusion-associated reactions (IAR), cerebrovascular complications and potential development of secondary autoimmunity, even years after the last administration.2 3 In this context, we analysed a large real-world prospective cohort of patients with MS in order to assess the potential impact of pretreatment on the efficacy and safety of ALEM infusion.
Methods
Patients
Between February 2014 and April 2018, adult patients with active MS, according to 2010 revised McDonald criteria,11 who were considered eligible for treatment with ALEM, based on the most recent prescription criteria, were enrolled in our prospective PROGRAMMS cohort (NCT04082260). ALEM infusions were administered at two tertiary MS centres (Muenster & Essen, Germany). All patients received ALEM according to the manufacturer’s guidelines. Exclusion criteria were: any progressive form of MS, inability to undergo MRI examination, presence of autoimmune disorders other than MS, systemic disease that interfere either with disability due to MS or the safety profile of ALEM; a more detailed description has been previously published.12
Outcome measurements
Epidemiological data were validated on screening and baseline infusion including disease and treatment history as well as determination of smoker-status since this was previously identified as possible risk factor for development of secondary autoimmunity.13 IARs were documented and graded according to the common terminology criteria for adverse events (CTCAE). Patients were evaluated every 3 months with standardised neurological examinations by two trained neurologists per site; the level of disability was scored by using the Expanded Disability Status Scale (EDSS). The occurrences of relapses, including their exact date of onset, the performed treatment and symptoms characterising the affected functional system, were recorded. Furthermore, localisation of symptomatic inflammation in the central nervous system (CNS) was determined by clinical evaluation and MRI data from semiannual scans. In the case of multifocal relapses, the localisation driving the relapse-related disability increase was counted.
In this study, we included only patients with a documented follow-up of at least 1 year following the second ALEM infusion course and with complete data on previous DMT, including treatment duration, date and reason for treatment cessation. The washout duration was defined as time from last drug intake to the first ALEM infusion.
Statistical analysis
Baseline parameters in our cohort were assessed using descriptive statistics. Patients receiving basic treatments (beta-IFN, DMF, glatiramer acetate, teriflunomide) were merged into one group (referred to as ‘basic’ group) since baseline characteristics (online supplemental table S1) and outcomes (online supplemental figure S1) were similar. For analysis of efficacy and safety outcomes, we used the Kaplan-Meier method and the Cox proportional hazard model. We defined ‘time to first clinical relapse’, ‘time to first confirmed worsening of disability’ and ‘time to first manifestation of secondary autoimmune disease (SAD)’ (each measured in months since baseline infusion) as meaningful outcome parameters for the regression analysis. Our regression models included the following covariates with an enter method: sex, age (above vs below median, since data were not normally distributed in naïve patients), annualised relapse rate at baseline, baseline-EDSS, disease duration since onset and last previous DMT. Multivariate HRs are stated throughout the manuscript. Worsening of disability was considered clinically relevant if two independent clinical assessments 3 months apart indicated an increase of the EDSS as follows: +1.5 points (baseline=0.0), +1.0 point (baseline=1.0–4.0), +0.5 points (baseline ≥4.5). To determine the progression to secondary progressive MS, Lorscheider criteria14 were used. Further analyses were carried out using Fisher’s exact test or the χ² test for categorical variables and Wilcoxon’s paired rank-sum test for continuous variables. A p<0.05 was considered significant. All analyses were considered exploratory. The analysis was carried out using SPSS Statistics 27.
Supplemental material
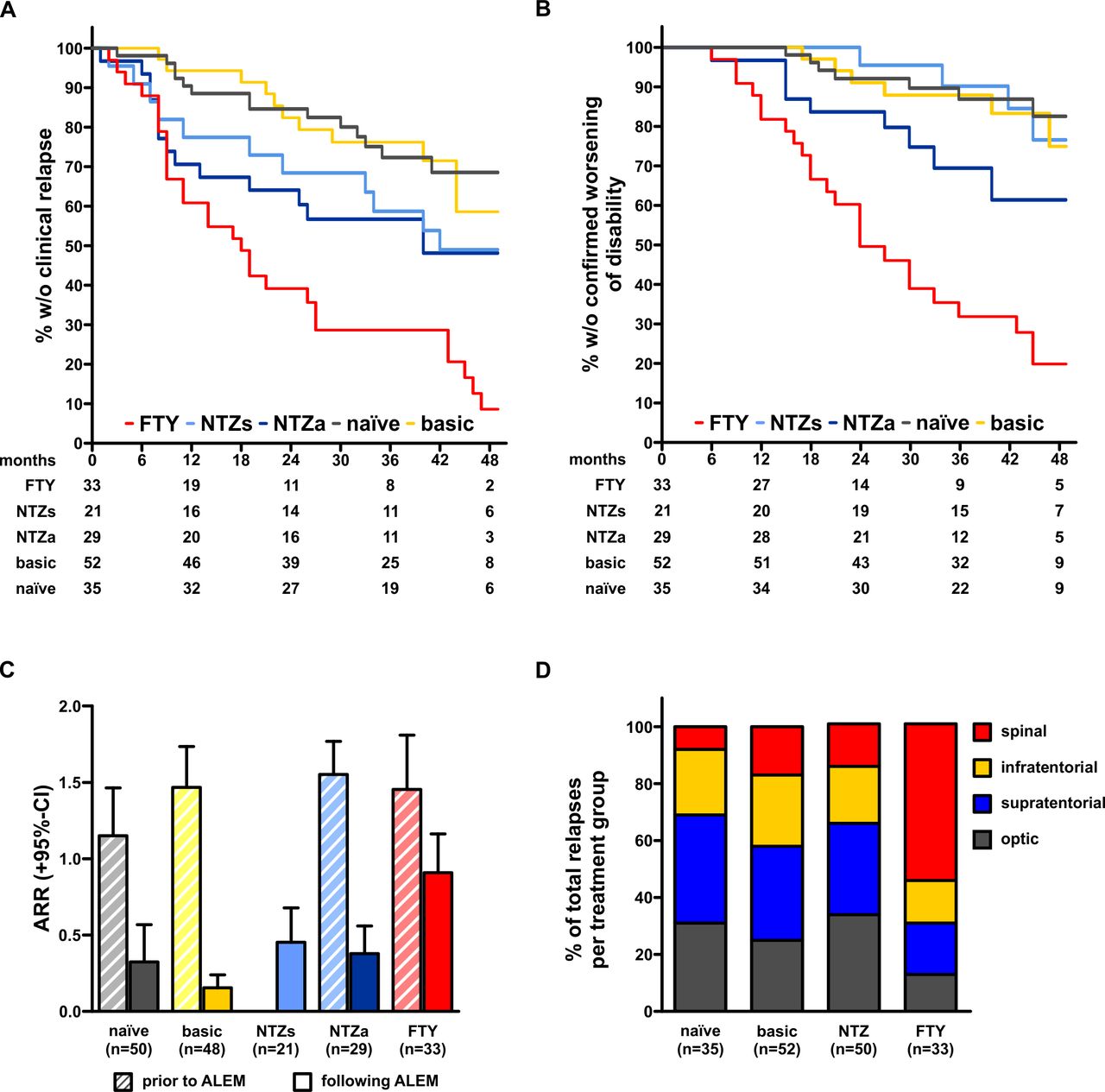
Analysis of efficacy outcomes in the PROGRAMMS cohort. (A) Kaplan-Meier plot depicting time to first clinical relapse of patients stratified to last previous disease-modifying treatment. Numbers below the x-axis indicate patients at risk at the respective time point. (B) Kaplan-Meier plot depicting time to first 3 months confirmed worsening of disability. (C) Analysis of annualised relapse rates 1 year prior to ALEM induction (left, striped bars) and in year 1 and 2 following induction (right bars) divided by last previous disease-modifying treatment. Data are shown as mean+95% CI. Numbers above bars indicate significance levels determined using the Wilcoxon-paired rank-sum test. (D) Depiction of relative relapse distribution in different treatment groups. ALEM, alemtuzumab; FTY, fingolimod; NTZa, natalizumab (previously active); NTZs, natalizumab (previously stable).
Distribution of baseline data in the PROGRAMMS cohort
Results
Patients
We identified 170 patients who were treatment naïve or previously failed to respond to NTZ, FTY, IFN or DMF, and were treated with at least two courses of ALEM. In total, data from 2425/2498 (97.1%) scheduled visits were available. All patients had received their previous DMT for more than 6 months. Majority of patients (n=108) were switched to ALEM because of the occurrence of disease activity, although 21 NTZ-treated patients were switched because of the increased risk for developing progressive multifocal leukoencephalopathy (PML), while having experienced stable disease.
Baseline clinical and demographic features were similar, among treatments groups. However, there were higher proportions of male patients in the ‘naïve’ and ‘basic’ groups. In addition, treatment naïve patients were younger and had shorter disease duration at ALEM commencement (table 1).
Relapses and disability worsening
In the total population, 78 patients (45.9%) experienced at least one relapse within the observation period; this occurred in three patients (1.8%) within the first 3 months and in 34 patients (20.0%) within 1 year following the ALEM infusion. We evaluated whether treatment effectives of ALEM depended on the number of previously administered DMT. Patients who received ALEM as third-line treatment, had a significantly increased hazard for relapses (HR 2.651, 95% CI 1.279 to 5.497; p=0.009), compared with those treated withALEM, as first or second line therapy (online supplemental table S2A). In the same group, a similar trend was observed for increased risk of disability worsening (HR 2.527, 95% CI 0.961 to 6.649; p=0.060; online supplemental table S2B). No relevant differences between patients having received ALEM as first-line or second-line treatment were noted.
Regression model for analysing time to first clinical relapse
Next, we investigated the response to ALEM based on the last previously used DMT. Among all treatments, the exposure to FTY was found to have the most significant association with an increased hazard of experiencing a clinical relapse, compared with treatment naïve and basic groups (figure 1A). The multivariate model confirmed that the previous use of FTY exerted the strongest predictive effect for an increased risk of relapses following ALEM infusion (HR 5.420, 95% CI 2.520 to 11.660; p<0.001) (table 2).
Additionally, the model identified the relapse rate at baseline (HR 1.460, 95% CI 1.098 to 1.940; p=0.009) and the previous exposure to NTZ, as significant predictors of the occurrence of clinical relapses.
Among previously NTZ-treated patients, we evaluated separately those who switched to ALEM because of increased PML risk while having been clinically stable(‘stable’ patients) and those who did because of ongoing disease activity (‘active’ patients). Compared with naïve patients, we observed higher hazards for relapses as well among ‘active’ patients (HR 3.888, 95% CI 1.375 to 10.990; p=0.010) as also among ‘stable’ patients (HR 2.732, 95% CI 1.138 to 6.560; p=0.025) (table 2).
Similar results were found when assessing the hazard for developing 3 months confirmed worsening of disability, which was significantly higher among patients previously treated with FTY (HR 7.676, 95% CI 2.870 to 20.53; p<0.001), compared with naïve patients. However, in the multivariate regression model, no further covariates, including the previous exposure to NTZ, were shown to affect the disability outcome (figure 1B and table 3).
Regression model for analysing time to confirmed worsening of disability
We also evaluated the number of total relapses during the first 2 years following ALEM induction (and thereby prior to any additional courses that were eventually administered). We found a strong reduction of the annualised relapse rate, compared with baseline, among treatment naïve patients (0.33 vs 1.15; p=0.004) and among those who had received basic treatment (0.16 vs 1.47; p<0.001). Similar trend was observed among NTZ-pretreated patients with previously stable disease (0 vs 0.45; p<0.001), and among previously active NTZ-treated patients (0.38 vs 1.55; p<0.001). In the FTY pretreatment group, following ALEM there was smaller yet significant reduction of the relapse rate within first 2 years (0.90 vs 1.46; p=0.010; figure 1C).
The clinical and MRI-based analysis of relapse anatomic localisation showed similar distribution of symptoms, among patients who had received basic treatment, NTZ or who were treatment naïve, with most lesions located within the cerebral hemispheres or optic nerves. In contrast, among patients pretreated with FTY there was a high preponderance of spinal relapses (55%) following treatment with ALEM (figure 1D), although previous medical reports showed no trend in favour of spinal cord symptoms while receiving FTY.
We also evaluated the transition to secondary progressive MS and found a single patient who met the Lorscheider criteria at 46 months after the first administration of ALEM (previously treated with FTY).
Additional treatment courses
Because of ongoing disease activity, 4 patients in the basic treatment group, 6 patients pretreated with NTZ, 6 treatment naïve patients and 11 patients pretreated with FTY received a further course of ALEM after 32 median months from the baseline infusion (range: 24–58 months). In addition, two patients (one pretreated with NTZ and one pretreated with FTY) received a fourth course of ALEM at 45 and 55 months, respectively. Following retreatment, all patients in the basic and treatment naïve groups remained clinically stable, while three patients in the NTZ group experienced one further relapse within the observation period. In the FTY group, we observed seven further relapses, with one patient having experienced two relapses (and subsequently having received a fourth course).
SAD and IAR
Among 52 patients (30.6%), we observed SAD which are listed in table 4. Five patients developed two different autoimmune disorders (two patients: thrombocytopenia + Graves’ disease; two patients: vitiligo + Graves’ disease; one patient: idiopathic Castleman’s disease + Graves’ disease). Most of secondary autoimmunity presented with thyroid dysfunctions, although three patients experienced immune thrombocytopenia and a in a single case there was immune neutropenia. Furthermore, we observed four cases of vitiligo (previously published in ref. 15), one case of idiopathic multicentric Castleman’s disease (previously published in ref. 16) and two cases of autoimmune hepatitis.
Overview on observed secondary autoimmune disorders in the PROGRAMMS cohort
Similar to effectiveness analyses, we observed an increased hazard for development of secondary autoimmunity among patients treated with ALEM as third-line agent, compared with the first-line and second line groups (HR 2.850; 95% CI 1.060 to 7.424; p=0.038; online supplemental table S2C). In the multivariate model pretreatment with FTY was the only variable significantly influencing the risk of developing SAD, (HR 5.875; 95% CI 2.126 to 16.237; p<0.001), compared with naïve patients (figure 2A, for full regression model, see table 5). Notably, in the FTY pretreated group, the wash-out duration did not impact on the time to first manifestation of SAD (HR: 1.054 per additional day washout (95% CI 0.978 to 1.136; p=0.0172)). We also evaluated whether a history of smoking was associated with the development of SAD but could not find any significant effect; a history of smoking was recorded among 14 (27%) patients with SAD and among 25 patients (21%) without SAD (p=0.264).

{kind=link}
{kind=link}
Analysis of safety outcomes in the PROGRAMMS cohort. (A) Kaplan-Meier plot depicting time to onset of (first) secondary autoimmune disorder. (B) Analysis of infusion-associated reactions stratified by severity grade in patients who received their first course of ALEM. ALEM, alemtuzumab; CTCAE, common terminology criteria for adverse events; FTY, fingolimod; NTZ, natalizumab.
Regression model for analysing time to first secondary autoimmune disorder
Overall, 121 patients (71.1%) experienced IAR during their first course of ALEM, and 105 patients (61.2%) at the administration of the second course. In the majority of patients symptoms were mild, including fever, rash and tachycardia, each resolving without specific treatment (CTCAE°I–II). However, we also observed severe adverse events (CTCAE°III–IV) following the first course of ALEM, including temporary liver injury (one patient), symptomatic bradycardia (three patients), pneumonitis (three patients) and acalculous cholecystitis (six patients). We also reported laboratory changes indicative of gall bladder inflammation in the absence of symptoms in three of six patients during the second course. After stratification according to the last previous DMT, we found that patients who had received FTY were less likely to develop IAR during their first course of ALEM (figure 2B), whereas patients who had received NTZ were prone to develop such symptoms. Notably, the vast majority of severe IAR was observed in patients switching from NTZ (p<0.001).
Discussion
In this real-world study, we observed, among patients with MS treated with ALEM, different treatment responses, based on the previous use of DMT. Patients who had previously received either basic treatment or who were treatment naïve experienced outcomes for clinical relapses and disability progression rates comparable to those reported in randomised clinical trials.2 3 17 18 Furthermore, we confirmed previous data from the TREAT-MS study, showing a declining efficacy of ALEM proportionally to the number of previously used DMTs and therefore a better response following administration early in the disease course.5
We focused on the impact on the treatment response of the last previous DMT administered before ALEM. The analyses demonstrated that patients switching to ALEM from NTZ, compared with those previously on basic therapy or treatment naïve, were more likely to experience relapses. This was observed in both subgroups of previously NTZ-treated patients with or without previous ongoing disease activity. However, in both NTZ-treated subgroups, following the commencement of ALEM there was no significantly increased risk of disability worsening, compared with naïve patients unlike was previously observed in patients switching from NTZ to FTY.6 19 Overall, our data support the use of ALEM as a valuable treatment option for patients stopping NTZ.
Previous evidence regarding the effectiveness of ALEM in previously FTY-exposed patients remained conflicting. Whereas previous reports raised concerns regarding decreased effectiveness or even aggravation of disease courses within this special treatment sequence,8 other studies concluded that ALEM is an effective option in patients stopping FTY.9 10 Here, we showed that patients previously treated with FTY had worse response to ALEM, compared with other treatment groups, as they experienced a less pronounced reduction of annualised relapses rates compared with baseline as well as higher hazards for disability worsening and for the development of secondary autoimmunity. Yet, compared with their respective baseline, annualised relapse rates following ALEM induction were significantly lower in previous FTY-treated patients. We hence consider these patients as ‘suboptimal responders’ rather than as ‘non-responders’.
Previous reports have suggested ongoing lymphopenia as a possible risk factor for suboptimal treatment response. However, we observed normal lymphocyte counts (above 1200 cells/mm³) in all but three patients following FTY. Nonetheless, we assume that total blood lymphocyte counts might not depict differences in lymphocyte subsets and their respective tissue distribution. Lymphocytes are differentially affected by FTY and relevant populations might be retained in lymphoid tissues and thereby might be relatively spared from depletion.20 We can only speculate whether such phenomenon is the reason for the relative reduction of IAR in FTY pretreated patients, as such reactions are suggested to directly correlate with immune cell destruction.21 22
However, from the current dataset, we can neither confirm nor ultimately rule out that the extension of the wash-out period before commencing ALEM can positively affect outcome parameters (median washout in our cohort: 1.5 months vs 3.5 months in previous cohorts).
It is known that persistent T cell clones can become the source of homeostatic proliferation following ALEM treatment and previous data proved this as a pivotal step for manifestation of secondary autoimmunity.23 Although it has not been shown yet, we assume that similar mechanisms might underlie re-emerging disease activity, as distinct changes in the T cell receptor repertoire were visible prior to relapses in event-driven analyses.24
These hypotheses are further supported by the qualitative changes of relapse following FTY, which in large proportion localised in the spinal cord and indeed were persistent also in patients with longer washout durations. It has been shown that specific lymphocyte subsets have preferences for different parts of the CNS.25 Furthermore, the spinal cord involvement is an important driver of disability progression and is likely to underlie the observed increased risk of disability accumulation in our FTY pretreated patients.26
Besides differences in lymphocyte distribution and their accessibility for depletion, lots of other effects mediated by FTY have been described. These especially involve qualitative changes in the immune network, such as transcriptomic changes of CD4+ T cells,27 the modulation of T helper cell phenotype balances as well as the increase in regulatory T cell abundance28 and the increase and functional changes in regulatory B cells.29 30 We speculate that these effects interfered with ALEM-induced depletion and immune reconstitution in an unfavourable manner, resulting in increased risk for disability progression and for the development of secondary autoimmunity.
These long-lasting changes in the immune repertoire following induction with ALEM after FTY can also explain the absent association between washout duration and time to manifestation of SAD in our cohort. We assume that—unlike lymphocyte sequestration—the qualitative changes in the immune network following FTY treatment and subsequent impact of ALEM re-shape the immune system in an irreversible manner and that this cannot be overcome by re-exposition to ALEM.
Interestingly, core study data already indicated that the risk of developing SAD is mostly defined by the first course of ALEM with only minor changes in risk exerted by further courses.31
We did not observe specific patterns of autoimmunity in our treatment groups but we assume that other risk factors could define the organ direction of autoimmunity. We have previously shown that a genetic predisposition via human leucocyte-antigen haplotypes is visible in vitiligo patients and a high abundance of thyroid antibodies at baseline in patients with secondary thyroiditis.15 32 Additionally, the identification of a family history of autoimmunity or a history of smoking as risk factors for development of secondary autoimmunity corroborate the concept of dormant autoimmunity being unravelled by ALEM.13
We are aware that the absence of randomisation and a potential sample bias at our tertiary centres represent our study’s limitations. However, we should not expect randomised clinical trials with designs capable of evaluating hypotheses such as ours; such limitations have been repeatedly noted.33 Consequently, real-world longitudinal studies like PROGRAMMS remain invaluable for determining a definite safety and efficacy profile.
Although the efficacy of FTY has been proven in various clinical trials, such as FREEDOMS, TRANSFORMS or most recently PARADIGMS,34–36 our data indicate FTY pre-treatment as a risk factor for suboptimal therapeutic response to ALEM and for developing secondary autoimmunity. Additionally, our data provide an interesting insight into the complex interaction between immune cell distribution and qualitative immune cell function for treatment success in patients with MS and how immunomodulatory treatment persistently modifies this interaction.
Data availability statement
Data are available on reasonable request. Anonymised data will be shared on reasonable request from qualified investigators.
Ethics statements
Ethics approval
PROGRAMMS adheres to the Declaration of Helsinki, and ethical approval was given by local authorities (Institutional review board of the Medical Council Westphalia-Lippe, 2014–398 f-S).
Acknowledgments
We would like to thank Anna Lammerskitten, Sarah Niesner, Claudia Schwering, Carolin Risau, Karla Musiolik, Denise Putzer and Susanne Keeren for their assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SP and TR contributed equally.
HW and SGM contributed equally.
Contributors SP: study concept and design, acquisition of data, analysis of data, writing of the manuscript. TR: analysis of data, critical revision of the manuscript for intellectual content. RP: acquisition of data, analysis of data, critical revision of the manuscript for intellectual content. LR: analysis of data, critical revision of the manuscript for intellectual content. CK: acquisition of data, critical revision of the manuscript for intellectual content. MP: acquisition of data, critical revision of the manuscript for intellectual content. BW: critical revision of the manuscript for intellectual content. LK: critical revision of the manuscript for intellectual content. CK: critical revision of the manuscript for intellectual content. AS: critical revision of the manuscript for intellectual content. HW: study concept and design, critical revision of the manuscript for intellectual content. SGM: study concept and design, critical revision of the manuscript for intellectual content.
Funding The study was financially supported by Sanofi Genzyme (ALAIN01 to TR and SGM) and the Competence Network Multiple Sclerosis (01GI1603D, PROGRAMMS to HW and SGM).
Competing interests SP: received travel grants from Sanofi Genzyme and Merck Serono, lecturing honoraria from Sanofi Genzyme, Mylan Healthcare, and Biogen, and research support from Diamed, Merck Serono, and the German Multiple Sclerosis Society Northrhine-Westphalia. TR: received travel grants and financial research support from Genzyme and Novartis and received honoraria for lecturing from Roche, Merck, Genzyme, Biogen and Teva. RP: received honoraria for lecturing and travel expenses for attending meetings from Alexion, Bayer Health Care, Biogen, Merck Serono, Mylan, Novartis, Roche, Sanofi-Genzyme, and Teva; has received research funding from Novartis. LR: received travel grants from Merck Serono and Sanofi-Genzyme. MP: received speaker honoraria and travel reimbursements from Novartis. CK: received travel grants from TEVA, compensation for serving on Scientific Advisory Boards for Novartis, and speaker honoraria from Biogen, Genzyme, and Merck Serono. BW: received grants from the German Ministry of Education and Research, Deutsche Forschungsgemeinschaft, Dietmar Hopp Foundation and Klaus Tschira Foundation, grants and personal fees from Merck Serono, Sanofi Genzyme, Novartis pharmaceuticals, and personal fees from Bayer, Biogen, Teva Pharma. LK: received compensation for serving on Scientific Advisory Boards for Genzyme, Janssen, Novartis, and Roche. She received speaker honoraria and travel support from Biogen, Genzyme, Merck Serono, Novartis, Roche and TEVA. She receives research support from the German Ministry for Education and Research, the German Research Foundation, the IZKF Münster, IMF Münster, Biogen, Novartis, and Merck Serono. CK: received honoraria for lecturing and travel expenses for attending meetings from Almirall, Merck Serono, Desitin, Sanofi-Genzyme, Biogen, Teva, Bayer, Novartis, Boehringer Ingelheim, Pfizer, Bristol Myers-Squibb, Daiichi Sankyo, Siemens, Eisai, Biotronik, Roche, Stago, Ever Pharma, CSL Behring, Mylan, MedDay Pharmaceuticals, Amgen. HW: received compensation for serving on Scientific Advisory Boards/Steering Committees for Bayer Healthcare, Biogen Idec, Sanofi Genzyme, Merck Serono, and Novartis. He received speaker honoraria and travel support from Bayer Vital GmbH, Bayer Schering AG, Biogen, CSL Behring, EMD Serono, Fresenius Medical Care, Genzyme, Merck Serono, Omniamed, Novartis, and Sanofi Aventis. He received compensation as a consultant from Biogen Idec, Merck Serono, Novartis, Roche, and Sanofi-Genzyme. HW also received research support from Bayer Healthcare, Bayer Vital, Biogen Idec, Merck Serono, Novartis, Sanofi Genzyme, Sanofi US and Teva. SGM: received honoraria for lecturing and travel expenses for attending meetings from Almirall, Amicus Therapeutics Germany, Bayer Health Care, Biogen, Celgene, Diamed, Genzyme, MedDay Pharmaceuticals, Merck Serono, Novartis, Novo Nordisk, ONO Pharma, Roche, Sanofi-Aventis, Chugai Pharma, QuintilesIMS, and Teva. His research is funded by the German Ministry for Education and Research (BMBF), Deutsche Forschungsgemeinschaft (DFG), Else Kröner Fresenius Foundation, German Academic Exchange Service, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, German Foundation Neurology and by Almirall, Amicus Therapeutics Germany, Biogen, Diamed, Fresenius Medical Care, Genzyme, Merck Serono, Novartis, ONO Pharma, Roche, and Teva.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Editorial commentary