Article Text

Abstract
Neurometabolic diseases are a group of individually rare but numerous and heterogeneous genetic diseases best known to paediatricians. The more recently reported adult forms may present with phenotypes strikingly different from paediatric ones and may mimic other more common neurological disorders in adults. Furthermore, unlike most neurogenetic diseases, many neurometabolic diseases are treatable, with both conservative and more recent innovative therapeutics. However, the phenotypical complexity of this group of diseases and the growing number of specialised biochemical tools account for a significant diagnostic delay and underdiagnosis. We reviewed all series and case reports of patients with a confirmed neurometabolic disease and a neurological onset after the age of 10 years, with a focus on the 36 treatable ones, and classified these diseases according to their most relevant clinical manifestations. The biochemical diagnostic approach of neurometabolic diseases lays on the use of numerous tests studying a set of metabolites, an enzymatic activity or the function of a given pathway; and therapeutic options aim to restore the enzyme activity or metabolic function, limit the accumulation of toxic substrates or substitute the deficient products. A quick diagnosis of a treatable neurometabolic disease can have a major impact on patients, leading to the stabilisation of the disease and cease of repeated diagnostic investigations, and allowing for familial screening. For the aforementioned, in addition to an exhaustive and clinically meaningful review of these diseases, we propose a simplified diagnostic approach for the neurologist with the aim to help determine when to suspect a neurometabolic disease and how to proceed in a rational manner. We also discuss the place of next-generation sequencing technologies in the diagnostic process, for which deep phenotyping of patients (both clinical and biochemical) is necessary for improving their diagnostic yield.
- neurogenetics
- metabolic disease
- clinical neurology
- mitochondrial disorders
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Inborn errors of metabolism with neurological manifestations, or neurometabolic diseases, are a group of heterogeneous genetic disorders that share in common the alteration of specific aspects of the cellular metabolism, ultimately leading to disease. Often first described by paediatricians, the more recently reported adult-onset forms have phenotypes sometimes considerably different from paediatric ones, which may mimic other more common neurological disorders in adults, thus justifying a specific approach.1–3 Adult-onset neurometabolic diseases are individually rare, numerous, heterogeneous and frequently show complex clinical presentations. These reasons, in addition to the myriad of specialised biochemical diagnostic tools available,4 account for a significant diagnostic delay and underdiagnosis.5 However, unlike many other neurogenetic diseases, a substantial part of neurometabolic diseases can be successfully treated, with both conservative and more recently approved innovative therapeutics. Early recognition and diagnosis of a treatable neurometabolic disease can have a major impact for patients, leading to the stabilisation of the disease or even the regression of some signs and symptoms, halting unnecessary diagnostic investigations, and allowing for family screening and treatment of presymptomatic carriers.
For all of the aforementioned, an overview of adult-onset neurometabolic diseases will be outlined, from important general considerations to phenotypical descriptions focused on treatable diseases. Furthermore, a simplified diagnostic approach for the adult neurologist will be presented, with the aim to help determine when to suspect a neurometabolic disease and how to further proceed in a rational manner.
Methods
Search strategy and selection criteria
Neurometabolic diseases are a group of genetic disorders that share in common the alteration of the cellular metabolism, ultimately leading to disease including neurological manifestations. This definition has its limitations, given that many neurological diseases may be associated to changes in cellular metabolism that participate in some degree to the pathogenesis of the disease. Within the aim of a pragmatic clinical approach, this review will consider as a neurometabolic disease those that can be either diagnosed on characteristic biochemical abnormalities, indicating an alteration of an specific metabolic pathway, and/or those that may respond to treatments aimed at correcting a given metabolic dysfunction.6 In order to review all adult-onset neurometabolic diseases, we first searched PubMed for article abstracts published in English, French and Spanish using the search terms “inborn errors of metabolism”, “metabolic disease” and “neurometabolic disease”, and reviewed the book by Hollak and Lachmann in 20167 to assemble all adult-onset neurometabolic diseases, including patients from any ethnic origin (see online supplemental material 1). Furthermore, we consider as ‘adult onset’ those patients with a genetically confirmed neurometabolic disease and an onset of neurological symptoms after 10 years since patients with a given neurometabolic disease and an onset of progressive neurological symptoms after this age usually present a rather homogeneous phenotype, which differs from earlier-onset forms.8–10 We did not exclude as adult onset those patients with subtle and non-progressive symptoms before the age of 10 years of age that could go overlooked, such as mild intellectual disability.
Supplemental material
Subsequently, we undertook a focused search in PubMed for articles in those same languages, using each previously identified adult-onset neurometabolic disease as a search term, with the aim to refine the adult-onset neurometabolic diseases selection and to identify those where a disease-modifying therapy is currently available (figure 1 and online supplemental material 2). We collected all clinical, MRI, biochemical and therapy data concerning these diseases. Although numerous and different classifications exist, these treatable adult-onset neurometabolic diseases were classified and presented in a clinically relevant manner, meaning that disorders with similar clinical characteristics and common biochemical tests were grouped together.
Supplemental material
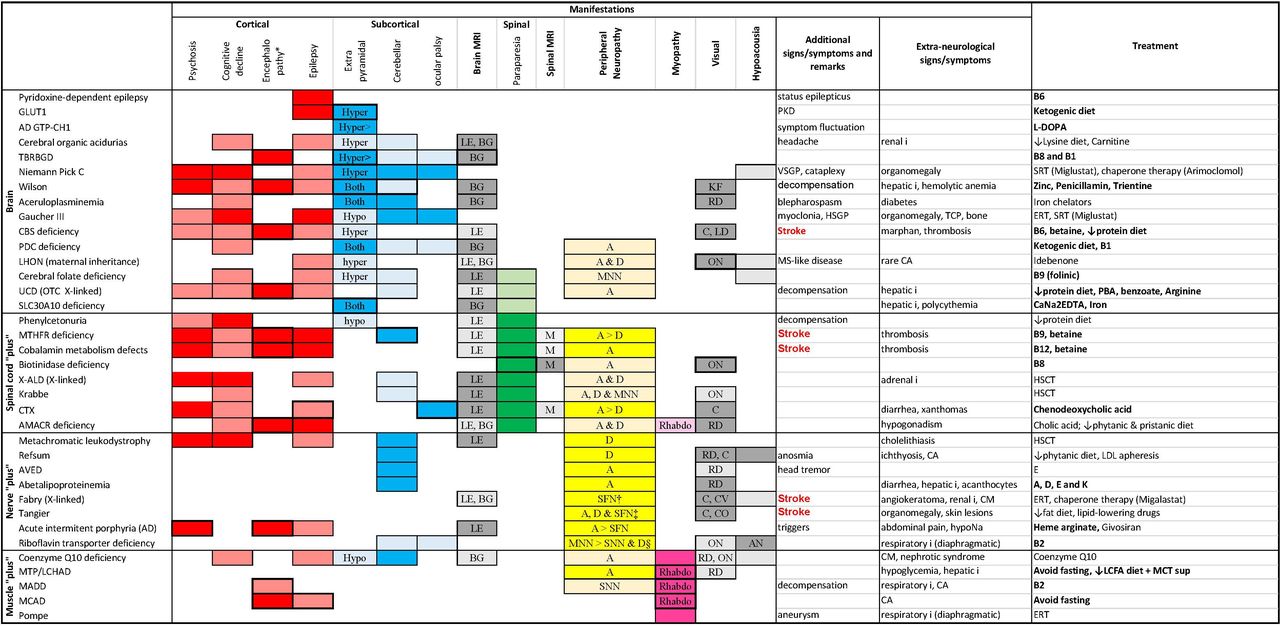
Neurological and extraneurological manifestations of treatable adult-onset neurometabolic diseases. Neurometabolic diseases are classified according to their neurological systematisation: brain: encephalic involvement; spinal cord ‘plus’: associated spinal cord involvement; nerve plus: associated nerve involvement; muscle plus: associated muscle involvement. The boxes have been coloured according to the topography of the manifestation: red for cortical, blue for subcortical, green for spinal cord, yellow for peripheral nerve and pink for muscle manifestations. Dark colours correspond to ‘major’ manifestations, whereas dull colours correspond to ‘minor’ manifestations (see text). When the box is framed in bold, it means that the manifestation in question may be acute/subacute in presentation. Extrapyramidal involvement is hypokinetic when it corresponds to parkinsonism and hyperkinetic in the case of dystonia chorea or ballismus. Cobalamin metabolism defect: predominantly clbC and cblB deficiency. *Acute or subacute change in mental status (confusion or coma). †Compressive neuropathy. ‡Pseudo-syringomyelic syndrome, multifocal demyelinating polyneuropathy with conduction blocks. §Cranial neuropathy. The authors have created and have permission to use the image. A, axonal; AD GTP-CH1,autosomal dominant GTP cyclohydrolase 1; AMACR, alpha-methylacyl-CoA racemase; AN, auditory neuropathy; AVED, ataxia due to vitamin E deficiency; BG, basal ganglia signal abnormality; C, cataract; CA, cardiac arrythmia; CBS, cystathionine beta-synthase; CM, cardiomyopathy; CO,corneal opacities; CTX, cerebrotendinous xanthomatosis; CV, cornea verticillata; D, demyelinating; ERT, enzyme replacement therapy; HSCT, haematopoietic stem cell transplantation; HSGP, horizontal supranuclear gaze palsy; hyper, hyperkinetic; hypo, hypokinetic; I, insufficiency; KF, Keyser-Fleisher ring; LCFA, long-chain fatty acid; LCHAD, long-chain L-3 hydroxyacyl-CoA dehydrogenase deficiency; LD, lens dislocation; LDL, low-density lipoprotein; LE, leucoencephalopathy; LHON, Leber hereditary optic neuropathy; M, myelitis (spinal MRI T2 hyperintensities); MADD,multiple acyl-CoA dehydrogenase deficiency; MCAD, medium-chain acyl-coenzyme A dehydrogenase deficiency; MCT, medium-chain triglyceride; MNN, motor neuronopathy; MS, multiple sclerosis; MTHFR, methylenetetrahydrofolate reductase; MTP, mitochondrial trifunctional protein deficiency; ON, optic neuropathy; OTC, ornithine transcarbamylase; PBA, phenylbutyrate; PDC, pyruvate dehydrogenase complex; PKD, paroxysmal kinesigenic dyskinesia; RD, retinal dystrophy; Rhabdo, rhabdomyolysis; SFN, small-fibre neuropathy; SNN, sensory neuronopathy; SRT, substrate reduction therapy; TBRBGD, thiamin and biotin responsive basal ganglia disease; TCP, thrombocytopenia; UCD, urea cycle disorder; VSGP, vertical supranuclear gaze palsy; X-ALD, X-linked adrenoleucodystrophy.
REVIEW
Key concepts in neurometabolic diseases
Pathophysiology
Neurometabolic diseases are often due to an enzyme deficiency or dysfunction, potentially causing substrate accumulation upstream of the enzymatic block and a lack of downstream product synthesis (figure 2). If the accumulation of substrates is toxic at abnormally high concentrations, it may be responsible for clinical manifestations.6 These manifestations may be acute, as in ammonium accumulation in urea cycle disorders (UCDs)11 or haeme precursors’ accumulation in acute intermittent porphyria,12 and/or progressive, as in the case of lysosomal storage disorders (LSD).13 Furthermore, the toxic accumulation may also be due to a transport defect (as in Wilson and aceruloplasminaemia). In other neurometabolic diseases, it is the deficiency in a given enzymatic product that leads to disease, either directly, as in neurotransmitter synthesis disorders,14 or indirectly, due to impaired downstream cellular function like in pyruvate dehydrogenase complex (PDC) deficiency (in this case, in addition to lactic acidosis).15 Moreover, due to the complexity of metabolic networks, a deficiency in a given enzyme can also lead to less schematic metabolic derangements with both indirect increased toxic compounds as well as deficient ones. The observed enzyme deficiency is not always due to an anomaly of the enzyme itself, but it can also be explained by a defect in substrate or enzyme transport to the catabolic site (as in X-linked adrenoleucodystrophy (X-ALD) where peroxysomal transport of very long chain fatty acids is impaired)16; by a deficiency in a protein with the function of bringing in proximity the enzyme and its substrate (as in saposin deficiency, responsible for a metachromatic leucodystrophy with arylsulfatase normo-function)17; or by an alteration in the metabolism of a vitamin required for the synthesis of a cofactor essential for the correct functioning of the enzyme (as the impaired synthesis of the 5-methyltetrahydrofolate, the active form of folate occurring in methyltetrahydrofolate reductase (MTHFR) deficiency and leading to the subsequent altered methionine synthase activity responsible for hyperhomocysteinaemia and hypomethioninaemia).10

Schematic representation of the pathophysiology and potential therapeutic strategies in neurometabolic diseases. In blue squares are specific treatments within the three main therapeutic strategies and the diseases where the treatment is indicated. *Treatment is also a free-radical scavenger. **Treatment will also decrease substrate accumulation by downregulating its synthesis. The authors have created and have permission to use the image. A, substrate; AD GTP-CH1, autosomal dominant GTP cyclohydrolase 1; AMACR: alpha-methylacyl-CoA racemase; AVED, ataxia due to vitamin E deficiency; B, product; cblC, cobalamin C; CBS, cystathionine beta-synthase; CTX, cerebrotendinous xanthomatosis; E, enzyme; HSCT, haematopoietic stem cell transplantation; LCHAD, long-chain L-3 hydroxyacyl-CoA dehydrogenase; LHON, Leber hereditary optic neuropathy; MADD, multiple acyl-CoA dehydrogenase deficiency; MCAD, medium-chain acyl-coenzyme A dehydrogenase; MTHFR, methylenetetrahydrofolate reductase; MTP, mitochondrial trifunctional protein deficiency; OTC, ornithine transcarbamylase; PDC, pyruvate dehydrogenase complex; TBRBGD, thiamin and biotin responsive basal ganglia disease; X-ALD, X-linked adrenoleucodystrophy.
Biochemical tools
Compared with other neurogenetic diseases, the diagnostic approach in neurometabolic diseases relies in the use of very numerous biochemical tests.4 These may be the measurement of one or a set of metabolites through a single test (such as total homocysteine or amino acid chromatography, respectively), of an enzymatic activity or of a functional test such as the mitochondrial respiration. Those tests may be performed in different fluids or tissues (including blood, urine, CSF, muscle or cultured cells, mainly skin-derived fibroblasts). Some biochemical studies are broad and point towards an overall alteration of a metabolic pathway; thus, additional analysis may be needed to identify the specific location of the metabolic block. These should also help guide and/or interpret the genetic study, whether it is Sanger sequencing of a specific gene or next-generation sequencing (NGS) of a metabolically oriented panel of genes.18 For example, an abnormal profile of blood acylcarnitines is suggestive of an altered mitochondrial beta-oxidation of fatty acids, and the nature of the specific abnormal acylcarnitines can point toward a specific enzyme within this pathway. Other biochemical studies can only screen for a specific neurometabolic disease, such as blood cholestanol in cerebrotendinous xanthomatosis (CTX), or the different specific enzymatic activities in LSDs, among which only the subgroups of mucopolysaccharidoses and oligosaccharidoses, almost exclusively of paediatric onset, can be screened as a whole by analysing mucopolysaccharids and oligosaccharids in urine.19
Therapeutic strategies
Unlike most non-metabolic neurogenetic diseases, many neurometabolic diseases are treatable6; that is, the natural history of the disease can be modified by a specific therapeutic intervention. Some innovative treatments have been very recently proved to be efficient, and many therapeutic trials are ongoing in the field of neurometabolic diseases. Several and sometimes complementary therapeutic approaches exist (figure 2):
Restoration of a minimal enzymatic activity. It can be achieved by iterative intravenous infusion of the deficient enzyme as in Fabry and Pompe disease, known as enzyme replacement therapy (ERT).20 21 However, ERT is frequently unable to cross the blood–brain barrier and therefore has little impact on neurological symptoms. Various strategies are currently being studied to overcome this caveat, and recently, the intraventricular administration of ERT has been shown to be effective in neuronal ceroid lipofuscinosis type 2.22 Depending on the neurometabolic disease, increasing the enzyme activity may be possible through other strategies than ERT: (1) the administration of a high-dose vitamin or an enzymatic cofactor, such as vitamin B6 (pyridoxine) in certain forms of classic homocystinuria (cystathionine beta-synthase deficiency)23; (2) the administration of a molecule with a chaperone effect that will limit enzyme degradation, like migalastat in Fabry disease or arimoclomol in Niemann-Pick C disease (that increases heat shock protein expression)24 25; (3) haematopoietic stem cell transplant of cells expressing a normal activity of the deficient enzyme, whether allogenous (classically used in X-ALD, Krabbe disease and metachromatic leucodystrophy)26–28 or ex vivo genetically modified autologous stem cells expressing the deficient enzyme in a supraphysiological manner (recent trials have demonstrated the efficacy of this latter strategy in children with X-ALD and metachromatic leucodystrophy)29 30; and (4) liver and/or kidney transplant with restricted indications in organic acidurias and UCDs. Without directly acting on enzyme activity, some treatments similarly aim to support the deficient metabolic function: this mainly concerns defects of energy metabolism (Glut1 deficiency, PDC deficiency and mitochondrial beta-oxidation disorders) for which specific ketogenic diets can restore energy production, bypassing the altered energetic pathway.
Limiting or decreasing the accumulation of substrates upstream of the enzymatic block. This can be achieved by a specific diet or regime, such as the limitation of protein intake in UCDs and the consequent reduction of the ammonemia11; by a detoxifying drug which binds to these substrates and limit their toxicity or facilitate their elimination, as is the case for sodium benzoate in UCDs31; or, still in UCDs, by extrarenal depuration to decrease ammoniaemia in the context of severe disease decompensation. In LSDs, drugs that inhibit the endogenous synthesis of these accumulated substrates can be used, which are known as substrate reduction therapy, like miglustat in Niemann-Pick C disease, which inhibits the ganglioside synthesis pathway.32 Similarly, a novel RNA interference therapeutic targeting hepatic delta-aminolevulinate synthetase 1 messenger RNA prevents the accumulation of haeme precursors in acute intermittent porphyria.33
The supply or substitution of the deficient product downstream of the deficient enzymatic reaction, such as L-dopa supplementation in autosomal dominant GTP cyclohydrolase I deficiency, also known as Segawa’s disease or dopa-sensitive dystonia.34
When to suspect a neurometabolic disease in adults and how to proceed
As mentioned previously, early recognition and diagnosis of a treatable neurometabolic disease is particularly important. We will follow the algorithm presented in figure 3, which is the result of the exhaustive review of the literature added to the clinical experience of the authors, and should therefore be interpreted as such as it will not replace clinical reasoning for each individual patient. In practice, the adult neurologist can encounter three situations.
Characteristic presentation of a given neurometabolic disease

Recommended algorithm to identify and evaluate adult patients with suspected neurometabolic diseases. *Movement disorders include extrapyramidal manifestations as well as isolated ataxia. **Genetic hyperhomocystinaemias include CBS deficiency, MTHFR deficiency and cobalamin metabolism defects. ***In parallel with diagnostic NGS if available or followed by confirmatory genetic testing if positive diagnostic work-up. The authors have created and have permission to use the image. CBS, cystathionine beta-synthase; CSF, cerebrospinal fluid; CTX, cerebrotendinous xanthomatosis; DWI, diffusion-weighted imaging; EMNG, electromyoneurography; GAG, glycosaminoglycan; HyperHcy, hyperhomocysteinaemia; LCHAD, long-chain L-3 hydroxyacyl-CoA dehydrogenase; LHON, Leber hereditary optic neuropathy; MADD, multiple acyl-CoA dehydrogenase deficiency; MTP, mitochondrial trifunctional protein deficiency; NGS, next-generation-sequencing; NP-C, Niemann-Pick type C; OS, oligosaccharides; PDE, pyridoxine-dependent epilepsy; UCD, urea cycle disorder; X-ALD, X-linked adrenoleucodystrophy; VLCFA, very long chain fatty acids.
In some situations, the presenting phenotype may be immediately suggestive of a given adult-onset neurometabolic disease (as described in figure 1 for treatable diseases): the specific biochemical and/or genetic test should be performed directly.
Genetic presentation or atypical acquired presentation
More frequently, the clinical picture suggests a genetic disease (family history of similar symptoms, parental consanguinity or a very chronic course), or a commonly acquired aetiology is suspected in the first place but there are atypical features leading to consider other less common disease mimics. In these situations, a neurometabolic aetiology should be considered if some of the following ‘red flags’ are present:
Multiple neurological manifestations, either involving several different neuroanatomical structures (eg, the association of epilepsy, indicating supratentorial disfunction, and cerebellar ataxia, indicating infratentorial involvement), or associated extraneurological signs and symptoms (visual, auditory, cardiac, hepatobiliary or endocrine). This is usually the case for most neurometabolic diseases. However, some more specific features can be found in particular subgroups of adult-onset neurometabolic diseases: in mitochondrial diseases (progressive external ophthalmoplegia, ptosis, stroke-like episodes, sensory neuronopathy, optic neuropathy, retinopathy, cataracts, sensorineural deafness and diabetes),35 in LSDs (supranuclear gaze palsy, hepatosplenomegaly, facial dysmorphia and osteoarticular deformities)13 and in peroxisomal disorders (demyelinating polyneuropathy, retinopathy, cataract, sensorineural deafness, facial dysmorphia and osteoarticular deformities).36
Acute or subacute neurological manifestations occurring in a context of basal metabolism modification and/or increased energy demand, such as weight loss, prolonged fasting, drastic modification of diet, surgery, infection or drugs. This can occur in aminoacidopathies,37 disorders of energy metabolism (including mitochondrial respiratory chain and beta-oxidation disorders),38 disorders of vitamin metabolism23 and porphyrias.12
Principal types of brain MRI findings (figure 4):
A demyelinating cerebral white matter disorder with confluent, bilateral and more or less symmetrical T2 hyperintensities, in the absence of vascular risk factors, can suggest certain LSDs (Krabbe disease with involvement of the cortico-spinal tracts,39 metachromatic leucodystrophy with frontal-predominant periventricular hyperintensities),40 a specific peroxisomal disorder: X-ALD (with contrast enhancement in the inflammatory phase of the disease),41 several ‘cerebral’ organic acidurias (with periventricular hyperintensities and possible involvement of the U-fibres),42 43 as well as other diseases (see figure 1 for treatable ones) including CTX (with involvement of the dentate nuclei or the peridentate cerebellar white matter).44 Furthermore, the presence of a bilateral and symmetrical often longitudinally extensive involvement of specific spinal cord tracts (such as subacute combined degeneration), associated or not with cerebral involvement, may also suggest a diagnosis of a treatable neurometabolic disease such as CTX, disorders of homocysteine remethylation (MTHFR deficiency and cobalamin C deficiency)45 and biotinidase deficiency.46
A bilateral and symmetrical involvement of the deep grey matter or basal ganglia. These abnormalities can be seen in several disorders of energy metabolism including mitochondrial respiratory chain disorders (producing a Leigh syndrome if associated with encephalopathy),47 PDC deficiency,48 and thiamin and biotin responsive basal ganglia disease (TBRBGD)49; in cerebral organic acidurias (associated or not with white matter T2 hyperintensities),42 and in Wilson’s disease.50
Frequently, the brain MRI may be completely normal, and this should not rule out a neurometabolic disease if other clinical and paraclinical findings are present.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
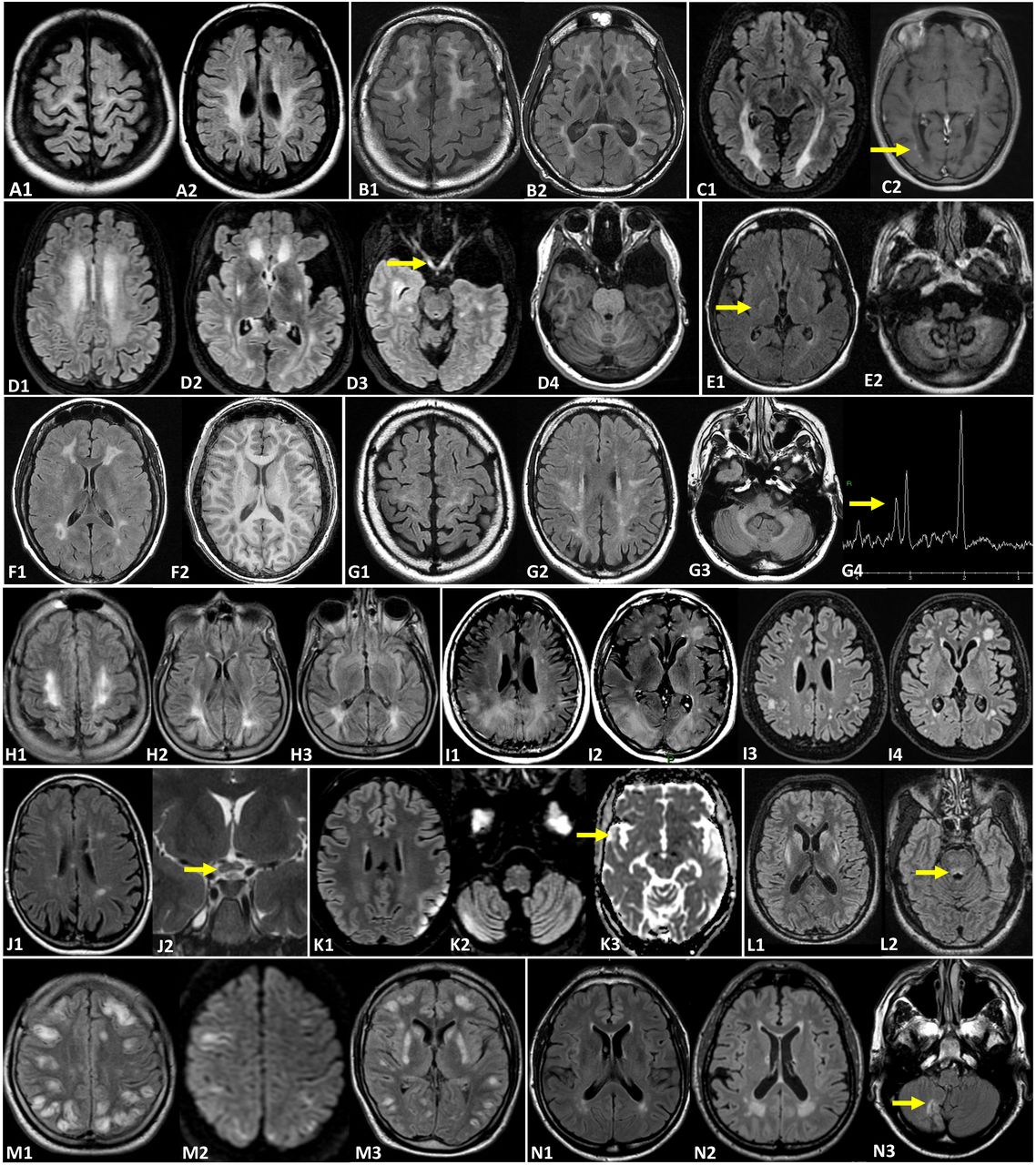
Brain MRI illustrating various treatable adult-onset neurometabolic diseases. All MRIs originate from adult patients (19–60 years old). Krabbe disease (A1–2): T2-FLAIR signal abnormalities involving the periventricular and deep white matter with frequent involvement of the corticospinal tract. Metachromatic leucodystrophy (B1–2): T2-FLAIR showing a diffuse leucoencephalopathy in particular around the frontal horns with sparing of subcortical U fibres leading to a ‘butterfly pattern’. X-linked adrenoleucodystrophy (C1–2): posterior leucoencephalopathy on T2-FLAIR (C1) and a peripheral border of active demyelination on gadolinium enhancement on T1W sequence (C2, arrow). Glutaric aciduria type 1 (D1–4): T2-FLAIR demonstrating a diffuse periventricular and deep white matter hyperintensity with lenticular (D2) and optic pathway (D3, arrow) signal abnormalities; enlargement of the convexity of subarachnoid spaces and sylvian fissures (D2) as well as bilateral temporal hypoplasia on T1W (D4). Cerebrotendinous xanthomatosis (E1–2): T2-FLAIR hyperintensity in the posterior limbs of both internal capsules (E1, arrow) and signal abnormalities within the dentate nuclei and the deep cerebellar white matter (E2). Methylenetetrahydrofolate reductase deficiency (F1–2): periventricular white matter signal abnormalities on T2-FLAIR (F1) and T1W (F2) sequences. Cerebrospinal fluid folate deficiency (G1–4): bilateral and symmetric leucoencephalopathy on T2-FLAIR sequences involving the supratentorial white matter and middle cerebellar peduncles. Spectroscopic analysis (G4) shows a rather low level of choline. Phenylketonuria (H1–3): periventricular and deep white matter leucoencephalopathy on T2-FLAIR predominantly located within posterior regions. Acute intermittent porphyria (I1–4) can sometimes be associated with posterior reversible encephalopathy syndrome (PRES) (I1–2). Complete disappearance of the PRES syndrome (I3–4) afterwards. Leber’s hereditary optic neuropathy ‘plus’ disease (J1–2): T2-FLAIR MS-like signal abnormalities (J1) and T2 central hyperintensity of the optic chiasma (J2, arrow). Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (K1–3): T2-FLAIR (K1–2) showing multifocal stroke-like cortical lesions in different stages of evolution involving multiple vascular territories. Increased signal on apparent diffusion coefficient MAP (K3, arrow) represents vasogenic rather than cytotoxic oedema. Wilson’s disease (L1–2): bilateral and symmetrical midbrain (L2, arrow), heads of the caudate and putamen T2-FLAIR hyperintensities. Thiamin and biotin responsive basal ganglia disease (TBRBGD) (M1–3): acute episode of a diffuse vasogenic oedema with numerous hyper T2-FLAIR (M1,3) and diffusion cortical lesions (M2). The heads of the caudate and putamen (M2) are also involved. Fabry’s disease (N1–N3): T2-FLAIR periventricular and deep white matter signal abnormalities. Compared with the baseline MRI (N1), the cerebral small vessel disease progresses on the 4-year follow-up MRI (N2) in a patient with no known cardiovascular disease. Ischaemic stroke sequelae of the posterior circulation (N3, arrow) can also be seen. The authors have created and have permission to use the image.T1W, T1 weighted.
In these situations, the neurologist should first search for subclinical signs that may guide the diagnosis: mainly a spinal MRI in the context of a pathological brain MRI, a nerve conduction study with electromyography, an ophthalmological evaluation to detect any corneal, lens, retinal and optic nerve alterations, and an audiological evaluation including an audiogram and auditory evoked potentials (that can be abnormal in the absence of audiogram abnormalities). Second, depending on the context, perform other complementary exams, including specific biochemical analysis, according to two possible parallel diagnostic strategies:
We will follow a “metabolic” strategy if the context is suggestive of a particular subgroup of neurometabolic diseases as previously mentioned: in this case diagnostic tests exploring the suspected metabolic pathways will be performed (see figure 3 and table 1).
In parallel, we will follow a “neurological” strategy where the biochemical diagnostic work up will be performed in accordance with the combination of neurological manifestations (see figure 1 for treatable diseases). Three main criteria are to be used to judge if a biochemical test can be useful for the patient: the consistency of the clinical presentation as a whole with the tested disease, the invasiveness and reliability (in terms of sensitivity and specificity) of the test, and the existence of a treatment. In this line figure 1 shows the different clinical manifestations of all potentially treatable adult-onset neurometabolic diseases categorised as major (if they are frequent or predominant within the clinical picture, and potentially be found isolated) or minor (if infrequent, even subclinical and rarely found isolated), as well as acute or chronic onset. Table 1 specifies the limitations of some screening tests. In the case of a treatable neurometabolic disease, it is probably legitimate to test not only if the presentation is reminiscent of the disease but also if it is merely compatible.
Key biochemical diagnostic tests for the diagnosis of treatable adult-onset neurometabolic diseases (clustered according to the explored metabolic pathways)
Unexplained isolated manifestation
Finally, some neurometabolic diseases may have an unspecific isolated neurological manifestation without the aforementioned clinical and paraclinical hints, compatible with either a genetic or acquired aetiology. For example, adrenomyeloneuropathy (the spinal form of X-ALD) can present as an isolated spastic paraparesis,51 and rarely, this is may also be the case for CTX.52 Niemann-Pick type C disease may present with isolated psychosis or a movement disorder.8 Certain neurometabolic diseases, like Tangier (possibly mimicking chronic idiopathic demyelinating polyneuropathy) or Pompe disease, may present with isolated neuromuscular signs and symptoms.53 54 In these cases, if the diagnosis is not obvious after first-line investigations, it may be useful to carry out a biochemical analysis of the possible neurometabolic diseases, in parallel to the relevant genetic study if necessary (eg, a ‘spastic paraplegia’ panel).
The recommended diagnostic approach is summarised in figure 3. If a biochemical test was to be positive, an evaluation by a specialist in the field should be considered to interpret the result within the clinical context and to guide the subsequent diagnostic process, whether further biochemical studies are needed or a genetic study can be undertaken. It should be noted that for some neurometabolic diseases, no reliable biochemical diagnostic test is available, and it is the genetic study alone that can confirm the disease. In these cases, it may be possible to perform a therapeutic test, of outmost importance in the case of neurometabolic diseases with possible acute/subacute presentations where benefit from early treatment can be dramatic. This is the case for TBRBGD and riboflavin transporter deficiency.23 Finally, the clinician should take into account the prevalence of individual neurometabolic diseases in their region, as well as the frequency of consanguinity and the presence of founding mutations.
Place for NGS gene technologies and metabolomics in the diagnosis of neurometabolic diseases
Neurometabolic diseases are predominantly, but not exclusively, single-gene recessive disorders. NGS technologies now allow for rapid and inexpensive large-scale genomic analysis of rare diseases.55 The first and most widespread technology is that of a panel of genes, allowing for the sequencing of hundreds of known genes implicated in neurometabolic diseases. If available, an alternative to gene panels is the sequencing of the entire set of protein-coding sequences or exome, known as wide exome sequencing or to directly sequence the entire 6 billion base pair human genome through wide genome sequencing. NGS has facilitated the detection of new gene–phenotype associations, expanding the clinical spectrum of already established Mendelian disorders.18 All of the aforementioned has increased our ability to correctly diagnose patients, has decreased diagnostic delay and reduced overall diagnostic expenses.56 Furthermore, NGS will also allow to detect genetic modifiers that could explain the phenotypical heterogeneity encountered within patients suffering from the same disease, even among siblings carrying the same pathogenic variants.57
However, the increased genetic resolution and complexity of the data comes with an increased detection of variants of unknown significance. This is why a combined approach with deep phenotyping of patients is needed. Concerning intellectual deficiency with a suspected metabolic origin, both clinical and biochemical phenotyping reached a high diagnostic yield of 68%,18 whereas similar exome studies without deep phenotyping usually result in a lower diagnostic yield of around 16%.58 To fit with this strategy, NGS, if easily available, could be performed in parallel to targeted biochemical phenotyping in patients with suspected neurometabolic diseases in the context of our proposed diagnostic approach. Complementary biochemical tests may also be needed once NGS results are available to help in the interpretation of variants of interest.
In addition, biochemical phenotyping could also be enlarged by metabolomics, establishing a comprehensive biochemical profile.59 The high-dimensional nature of the data amassed through metabolomics requires the use of dedicated preprocessing platforms and multivariate analysis or even machine learning tools to classify the findings.60 This untargeted approach has the potential to direct the diagnosis among varied and unspecific manifestations and to diminish the number of specific biochemical tests, thus reducing the overall cost.61 In addition, it could provide complementary biochemical data to assist in the interpretation of genetic variants from NGS gene studies.62 Although these metabolomics approaches are powerful, they have not yet been translated into clinical practice.
Conclusions
Adult neurometabolic diseases encompass a heterogeneous set of conditions for which we have presented a synthetic overview of the different phenotypes, especially for those with a currently available treatment, as well as a simplified diagnostic approach. Nevertheless, this practical knowledge is bound to change over the next years, with the identification of new neurometabolic diseases, the report of new phenotypes for known ones, the accessibility of untargeted diagnostic tools (genomics and metabolomics) and the discovery of new treatments for currently untreatable neurometabolic diseases. We hope that this review will increase awareness of this group of diseases and allow for an efficient and rational use of the biochemical tests available in the diagnosis of neurometabolic diseases, ultimately leading to prompt diagnosing and early treatment of patients.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Gorkaeulate
Contributors Concept and design, drafting of the manuscript: GF-E, CC YN. Administrative, technical, or material support: CC, NS and FL. Supervision: GF-E and YN. Acquisition, analysis or interpretation of data, critical revision of the manuscript for important intellectual content: all authors.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests YN reported grants from Takeda and Orchard therapeutics and has received honoraria from Sanofi Genzyme and Orphazyme. No other disclosures were reported.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.