Article Text

Abstract
Clinical case reports and prospective trials have demonstrated a reproducible benefit of hypothalamic-pituitary-adrenal (HPA) axis modulation on the rate of recovery from acute inflammatory central nervous system (CNS) demyelination. As a result, corticosteroid preparations and adrenocorticotrophic hormones are the current mainstays of therapy for the treatment of acute optic neuritis (AON) and acute demyelination in multiple sclerosis.
Despite facilitating the pace of recovery, HPA axis modulation and corticosteroids have failed to demonstrate long-term benefit on functional recovery. After AON, patients frequently report visual problems, motion perception difficulties and abnormal depth perception despite ‘normal’ (20/20) vision. In light of this disparity, the efficacy of these and other therapies for acute demyelination require re-evaluation using modern, high-precision paraclinical tools capable of monitoring tissue injury.
In no arena is this more amenable than AON, where a new array of tools in retinal imaging and electrophysiology has advanced our ability to measure the anatomic and functional consequences of optic nerve injury. As a result, AON provides a unique clinical model for evaluating the treatment response of the derivative elements of acute inflammatory CNS injury: demyelination, axonal injury and neuronal degeneration.
In this article, we examine current thinking on the mechanisms of immune injury in AON, discuss novel technologies for the assessment of optic nerve structure and function, and assess current and future treatment modalities. The primary aim is to develop a framework for rigorously evaluating interventions in AON and to assess their ability to preserve tissue architecture, re-establish normal physiology and restore optimal neurological function.
- NEUROOPHTHALMOLOGY
- MULTIPLE SCLEROSIS
- VISION
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Current treatment of acute optic neuritis
Corticosteroids
In 1961, Miller and colleagues demonstrated that patients with multiple sclerosis (MS), who were treated with corticotrophin recovered more quickly and completely from acute relapses than did patients treated with saline.1 A subsequent study of acute optic neuritis (AON) by Rawson and Liversedge2 demonstrated that a similar hastening of visual recovery was noted after acute retrobulbar neuritis; however, no significant difference between visual outcomes could be determined 12 months after the inception of visual symptoms. The emergence of intravenous methylprednisolone for the treatment of a range of immune-mediated disorders prompted its application for the treatment of acute MS exacerbations, including AON, whereby early benefits were noted regarding the course of clinical recovery.3
In 1992, the Optic Neuritis Treatment Trial (ONTT) provided the first comprehensive examination of the benefits of steroid therapy for AON in a large and representative patient cohort. In the ONTT, patients were randomised to receive placebo, oral (low-dose) prednisone (1 mg/kg/day for 14 days) or high-dose intravenous methylprednisolone (250 mg 4 times daily for 3 days), followed by oral prednisone (1 mg/kg/day for 11 days). At 6 months, colour vision and contrast sensitivity significantly improved in the methylprednisolone arm4; however, after 1 year, there was no significant difference between treated and untreated patients in any functional outcomes.5 Intravenous methylprednisolone was found to accelerate the rate of visual recovery over the first 15 days.4 Objective measures of optic nerve or retinal architecture were not available at the time of the ONTT; however, a subsequent study showed no effect of corticosteroids on optic nerve atrophy.6
In a subsequent analysis, patients randomised to receive treatment with high-dose intravenous methylprednisolone in conjunction with 11-day low-dose oral prednisone taper exhibited a significantly reduced risk of developing clinically definite MS (defined by the development of a second bona fide inflammatory demyelinating syndrome) over the subsequent 2 years.7 Notwithstanding this apparently noteworthy outcome, no significant disease-modifying effects were confirmed beyond the second year of ascertainment. Although the data from the ONTT have been reviewed rigorously, some have questioned the finding that intravenous steroids may produce a short-term benefit in delaying the onset of MS and that the risk of recurrent optic neuritis increases with the use of oral prednisone alone.8 Concerns include the lack of treatment blinding, post hoc analyses, small sample sizes, recoding of patient classifications and changes in the statistical assumptions. Despite the wealth of data gathered in the ONTT, critical questions regarding patient management remain unanswered. These include the following: (1) whether corticosteroid treatment is beneficial in patients whose symptom duration is longer than 8 days; (2) whether higher doses of corticosteroids are more effective than lower doses; (3) what is the optimal corticosteroid regimen; (4) whether the observed increased ON recurrence rate associated with oral prednisone also is observed in MS attacks and (5) whether high-dose methylprednisolone given periodically will improve the prognosis for patients with MS.
Intravenous immunoglobulin and plasma exchange
Intravenous immunoglobulin (IVIg) and plasma exchange (PLEX) also have been studied for the treatment of AON. Roed et al9 found no effect of IVIg on long-term visual function or on the latency of visual evoked potential (VEP) responses after AON. Further, IVIg did not improve visual function in patients with persistent vision loss after AON.10 Alternately, PLEX has demonstrated efficacy in the treatment of refractory AON11 and AON associated with neuromyelitis optica (NMO).12 Ruprecht et al11 observed a significant improvement in visual recovery following the institution of PLEX in cases of refractory AON; however, the rapid use of PLEX in this study may have masked any delayed benefit still to be derived from initial treatment with intravenous methylprednisolone. In fact, the authors noted that there is significant variability in the magnitude and tempo of efficacy derived from corticosteroid use among patients with AON. Indeed, a similar degree of variability can be observed in patients who derive clinical benefits from PLEX, either in isolation or following corticosteroid treatment.
A recent study evaluating the addition of PLEX to intravenous methylprednisolone in the acute treatment of NMO-associated AON showed significant improvements in high-contrast acuity, visual fields and temporal retinal nerve fibre layer (RNFL) thickness,12 but not low-contrast letter scores or colour vision. The early, first-line use of PLEX in the treatment of AON, however, has yet to be evaluated.
Erythropoietin
Systemic infusion of erythropoietin with and without methylprednisolone has demonstrated beneficial effects on retinal ganglion cell (RGC) function and survival in a rat model of experimental autoimmune encephalomyelitis.13 Erythropoietin administration increased protein levels of phospho-Akt, phospho-MAPK 1 and 2 and Bcl-2, indicating that activation of the Akt signalling pathway may be critical for limiting RGC apoptosis after AON. In combination with methylprednisolone, erythropoietin led to partial recovery of pattern-reversal VEPs and significantly improved flash electroretinograms (ERGs).13 Recently, a phase 2 clinical trial compared systemic erythropoietin with placebo in the treatment of AON in a small cohort of patients.14 Treatment with erythropoietin resulted in significant improvement in the average thickness of the peripapillary RNFL (as measured by optical coherence tomography (OCT)) and improved VEP latencies at week 16. Mean visual acuity showed a trend towards improvement after erythropoietin treatment. Given the inherent variability of optic nerve electrophysiology, the limitations of time-domain OCT, and the small study size, the paraclinical benefits observed with erythropoietin treatment will need to be confirmed in a multicentre prospective investigation.
Why a renewed focus on optic neuritis?
Novel technologies and an improved understanding of neuroinflammation have provided an ideal environment for renewed investigations into the early treatment for AON (table 1). Spectral domain OCT, scanning laser polarimetry (SLP), diffusion tensor imaging (DTI), multifocal VEPs (mfVEP) and the optic nerve head component (ONHC) of the multifocal ERG (mfERG) represent technical advancements in our ability to characterise precisely and objectively the complex architecture of the retina and optic nerve and correlate structural integrity with corresponding functional measures of visual system physiology.15–22 The application of these new tools to the investigation of structure-function derangements in AON will validate using the eye as a surrogate for studying the various mechanisms of central nervous system (CNS) injury in MS and provide a clinical paradigm to assay the potential neuroprotective and neurorestorative effects of therapeutic agents. These contentions have already been partly corroborated by a number of recent investigations that have provided seminal insights into the pathobiological underpinnings that drive structural and functional derangements in the visual system of patients with MS.
Investigations into the early treatment for AON
OCT/scanning laser polarimetry
OCT and SLP are optical imaging modalities that can measure the RNFL thickness. Already, multiple investigations have confirmed that both OCT and SLP have the capability to confirm significant thinning of this inner retinal layer composed of RGC axons and glial tubules formed by Muller cell processes. The RNFL axons are unmyelinated and derived from the ganglion cells of the macular retina, which become organised at the neural-retinal rim (the tissue between the papilla of the optic disc and the edge of the optic cup), beyond which point they are consolidated as the optic nerve (composed of about 1–1.2 million axons). As the optic nerve axons traverse the lamina cribrosa, they acquire myelin derived from CNS oligodendrocytes, with their corresponding electrical transmission properties transforming from slow membrane to highly rapid, saltatory conduction.17
OCT measures the interference spectrum of infrared light that has penetrated through the retina using a spectrometer and highspeed linescan camera. In spectral domain OCT, the ‘backscattered’ spectrum is Fourier transformed to obtain the magnitude and echo time delay of the light, thereby producing a set of axial images for analysis.23 Alternatively, SLP is an imaging technique that utilises the polarising birefringence properties of the RNFL to determine thickness of the retinal layers.24 The principal compositional element thought to contribute to RNFL birefringence is the intra-axonal microtubule. Because the SLP-derived RNFL thickness is calculated using the phase shift of polarised light, compensation for corneal birefringence is required and may be problematic in some individuals.24
Owing to their distinct technologies, OCT and SLP demonstrate RNFL oedema and thinning with varying efficacy. Specifically, OCT shows RNFL swelling (as water influences infrared light backscatter in OCT, whereas oedema is invisible to SLP because water is not a birefringent medium) better than SLP.25 Both technologies, however, appear equally efficient in measuring RNFL loss after acute inflammatory injury. Alternately, determining the true baseline RNFL thickness measure early in AON may be more accurate when utilising SLP, given that thickness measures are confounded by oedema in OCT. Despite the potential limitations of OCT in the acute setting of AON, the advent of high-precision retinal segmentation with spectral domain OCT (see below) has elucidated exciting data that suggest that detailed retinal analysis can yield baseline thickness measures that are not influenced by RNFL oedema and provide a ‘true’ baseline measure for the longitudinal analysis of putative neuroprotective therapeutic agents.22
The introduction of SLP and OCT in the eye clinic has greatly enhanced the appreciation for the timing and extent of axonal loss after retrobulbar AON. RNFL oedema as measured by OCT and SLP is far more common than noted on clinical examination.25 Oedema is evident in approximately 82% of affected optic nerves and remains evident in at least one quadrant in the majority of patients by 12 weeks.25 On average, as measured by OCT, patients with AON lose 22 µm more RNFL in their affected eye than in their unaffected eye in the 3–6 months after the inception of visual symptoms.26
To provide some perspective, the RNFL achieves a thickness of approximately 100 µm by 10 years of age, and over the subsequent 60 years of life, healthy individuals can expect to lose approximately 10 µm of thickness (corresponding to a rate of approximately 0.017% annually). After AON, RNFL loss is evident in the majority of individuals by 3 months,25 with nearly all patients showing segments of RNFL loss by 3 months.25 ,26 ,27 RNFL thinning as measured by OCT has been observed to correlate with multiple visual metrics: high-contrast acuity, low-contrast acuity, visual field and colour vision.26 ,28 ,29 For example, studies have demonstrated that a relevant loss of seven letters or more in low-contrast letter acuity predicts a corresponding reduction in the thickness of the RNFL in MS.28 The development and application of novel retinal segmentation algorithms for OCT have been used to sensitively and reproducibly measure anatomic compartments that reflect the extent of pathological and functional injury following AON. Specifically, the thickness of the ganglion cell and inner plexiform layers (GCL+IPL) facilitates measurement of ganglion cell loss after AON and correlates with visual function in affected individuals.18 ,22
MRI techniques
In contrast to OCT, conventional MRI has demonstrated mixed correlations between optic nerve atrophy and visual function after AON. In some investigations, the use of short-echo fluid-attenuated inversion recovery and T1-weighted spin echo imaging has revealed a significant relationship among the degree of optic nerve atrophy and visual acuity, VEP amplitudes and latencies.30 ,31 Across those studies that evaluated acute changes, the affected optic nerve was initially oedematous, with an increased mean cross-sectional area; later, optic atrophy developed. The administration of ‘high-dose’ intravenous methylprednisolone did not prevent or attenuate the subsequent development of optic nerve atrophy.32
Advanced MRI metrics, such as those derived with DTI and magnetisation transfer imaging ratios (MTR), have recently been adapted for orbital imaging.33 Traditionally, DTI and MTR methods have been employed infrequently for the purpose of orbital imaging, principally due to multiple and formidable technical challenges. In particular, the small size and mobile nature of the optic nerve, combined with confounding factors such as the signal intensity heterogeneity of the surrounding orbital structures (fat, bone and cerebrospinal fluid) mandate the application of rapid acquisition protocols in conjunction with high spatial resolution.34
In a non-conventional imaging study, MTR region of interest analysis showed a correlation with full-field VEP.35 In contrast, subsequent imaging analyses of the entire length of the optic nerve demonstrated a significant relationship with visual acuity but not with VEP latency changes.31 The MTR (a metric related to brain tissue integrity) in affected optic nerves declined slowly with a nadir of 240 days, a period that is longer than the standard interval for visual recovery.36 This may indicate that the window for treating AON may extend beyond the current clinical paradigm.
Recently, technical refinements in receiver coil characteristics, combined with high-field magnetic environments, have generated improved signal–to-noise ratios for DTI measures of axonal and myelin pathology in AON. Radial diffusivity has proven to be the most sensitive metric for differentiating the unaffected from affected optic nerves and correlates with visual recovery, electrophysiology (VEP latency and amplitude) and RNFL thickness (OCT).33 ,37 DTI, therefore, provides a novel, sensitive modality to complement RNFL structural injury in the evaluation of acute ON injury.
Electrophysiology
Electrophysiologic measures can reveal seminal features of axonal degeneration and inflammatory demyelination within the anterior visual system. Prolongation of the VEP P100 latency has long been used as a measure of conduction delay through the optic nerve and is a sensitive pathophysiological signature of demyelination.33 ,38 In contrast, a reduction in VEP amplitude can serve as a measure of axonal injury. As a summated response of multiple neuronal elements with differently oriented electrical dipoles, the full-field VEP is subject to significant limitations. First, the full-field VEP is dominated by the macular region and may not detect a significant fraction of ON cases with peripheral field loss. Next, full-field VEP may be hampered by changes in waveform architecture depending on the location of the optic nerve lesion or visual field loss.
Advancements in optics have introduced multifocal technology to VEP studies, and increased the ability to detect small changes within the central visual field.39 Full-field and mfVEP studies have demonstrated utility in predicting the magnitude of optic nerve injury and visual outcome in AON.39 ,40 The sensitivity of mfVEP may be enhanced further through the use of low-contrast pattern-reversal stimuli (analogous to the use of low-contrast letter acuity charts), allowing for the detection of mild residual injury or occult damage in the so-called ‘unaffected’ eye.16
The traditional ERG has had limited utility in the study of eyes in patients with MS. Nonetheless, the ONHC is a discrete late-response waveform of the ERG that can be used to detect electrophysiological changes in the context of acute AON. The ONHC is produced through a modified stimulus paradigm that includes global flash stimuli interleaved at specific intervals in the mfERG. The ONHC is thought to represent the transformation of slow membrane conduction to fast saltatory conduction, as axons traverse the lamina cribrosa and become myelinated.41 After AON, the ONHC waveforms are abolished and later recover, representing the transient effects of conduction block due to reversible demyelination. Frohman et al17 have demonstrated that eyes with previous optic neuritis in patients with MS exhibit changes or loss in the ONHC waveform that correlate with reduction in low-contrast letter acuity, RNFL thickness, visual field depression and amplitude loss and latency delay on mfVEP. Therefore, abnormalities of the ONHC response may provide a novel, additional pathophysiological signature of optic neuritis injury for acute treatment trials.
Biomarkers
Serum and plasma neurofilament levels, heavy (NfH) and light (NfL), are elevated in patients with AON, independent of the inflammatory mechanism.19 ,21 ,42 ,43 Supportive of a link between persistent vision loss and axonal degeneration, the levels of NfH and NfL have been observed to correlate with the extent of vision loss and the loss of retinal nerve fibre thickness following AON. Therefore, in addition to the aforementioned imaging and electrophysiological measures of optic nerve integrity and function, blood measures of NfH and NfL may provide additional information on neuronal loss and visual prognosis.
Neuroimmunology
AON lesions are rarely acquired for histopathological examination due to the limited nature of most injuries and the high probability for recovery. As a result, the composition of the inflammatory infiltrate in AON and the extent of glial and neuronal injury are inferred from that of acute CNS brain and spinal cord MS lesions. Demyelination in AON is presumed to be mediated by activation of endogenous microglia and a mixed inflammatory infiltrate consisting of T and B cells and peripheral macrophages. As in acute MS lesions, local expression of human leucocyte antigen (HLA) class II antigen, Th1 and Th17 proinflammatory T cells and axonal transections are evident.44 Tsoi et al45 noted pathological findings similar to those found in chronic active MS lesions in a 10-month-old ON lesions recovered at autopsy. These included myelin breakdown, infiltration and activation of macrophages and microglia, and gliosis. A detailed characterisation of the cellular infiltrate, measures of axonal transection, and local cytokine secretion were not performed.
The response of AON to interventions such as methylprednisolone, corticotrophin and PLEX implicate the combined action of cellular and humoral immune processes (table 2). The anti-inflammatory and immunosuppressive actions of corticosteroid administration and hypothalamic-pituitary-adrenal axis modulation are quite complex and act at multiple levels to reduce the acute inflammatory response.46 At the cellular level, intravenous corticosteroids reduce the number of circulating monocytes and lymphocytes by modulating cell apoptosis. At the cell surface, they reduce the expression of adhesion molecules and matrix metalloproteinase expression to lessen blood-brain barrier permeability. In addition, corticosteroids alter the transcription of proinflammatory and anti-inflammatory cytokines expressed by peripheral blood mononuclear cells. In a recent study, the proinflammatory cytokines IL-17A, IL-6 and IL-23p19 were downregulated by the administration of intravenous corticosteroids in patients with MS, whereas anti-inflammatory cytokines IL-10, TGF-β1 and IL-27p28 were upregulated.47 In other investigations, two groups noted enhanced T regulatory cell (Treg) function after the administration of intravenous corticosteroid treatment for acute MS relapse.48 ,49 These results suggest that corticosteroid therapy may restore impaired Treg function and reset the ratio of proinflammatory and anti-inflammatory cytokines after acute inflammatory demyelination. Although the potent anti-inflammatory effects of glucocorticoids have proven to be useful for increasing the rate of resolution of AON, it is possible that they have a negative impact on remyelination. In the cuprizone and experimental autoimmune encephalomyelitis animal models, high-dose glucocorticoids were found to inhibit remyelination,50 ,51 suggesting that there may be a need for AON therapies that can promote a more conducive environment for remyelination and neuronal recovery.
Clinical measures of optic nerve function and structure
PLEX is presumed to mediate a therapeutic effect, at least in part, through the removal of pathogenic humoral and plasma factors. Indeed, PLEX has been shown to benefit both patients with idiopathic AON and those with NMO AON.11 ,12 The utility of PLEX for the treatment of AON suggests that antibodies and/or proinflammatory serum components may facilitate optic nerve injury in AON.
Novel anti-inflammatory actions of adrenocorticotropin hormone
Recent studies have shown that α-melanocyte stimulating hormone and adrenocorticotropic hormone (ACTH) have anti-inflammatory effects in a number of models of acute CNS and ocular inflammation. ACTH is a member of the pro-opiomelanocortin-derived family of peptides called melanocortin peptides. Melanocortin peptides bind to G-protein coupled cell surface melanocortin receptors, of which five have been identified as melanocortin receptors 1–5 (MC1R–MC5R).52 The binding of ACTH to MC2R in the adrenal glands results in steroidogenesis. The other MCRs, however, are located throughout the body and are responsible for a variety of functions including direct, steroid-independent reduction of inflammation in the periphery and in the CNS (reviewed in52). Melanocortin peptides also have a unique role in maintaining immunological homoeostasis in the eye; they have been reported to induce a functionally important population of CD4+ regulatory T cells in the experimental autoimmune uveitis animal model.53 Additionally, there are reports that melanocortin peptides can have neurotrophic effects.54 Clinical studies with advanced physiological measures will need to be performed to elucidate whether these properties are relevant in optic neuritis.
Optic neuritis: alternative inflammatory injuries
A fraction of patients with AON experience this syndrome in the context of NMO, a demyelinating disorder directed against the aquaporin-4 (AQP4) water channel, with predilection for the optic nerves and spinal cord. Compared with that of MS-associated ON, vision loss secondary to ON in the context of NMO is typically severe (acuity loss, visual field loss),55 with a lower predilection towards significant recovery and a greater amount of axonal degeneration as measured by OCT.56 The greater magnitude in severity of the clinical syndrome, coupled with a worse prognosis for functional recovery in NMO-associated optic neuritis, may be a result of the targeted destruction of CNS and retinal astrocytes.
Among the broad diversity of CNS ‘housekeeping’ functions, astrocytes support neurotransmission and myelination, facilitate the clearance of extracellular potassium and water and facilitate nerve conduction. Further, astrocytes produce cytokines, such as platelet-derived growth factor, that stimulates the proliferation and differentiation of oligodendrocyte precursors into premyelinating oligodendrocytes.57 Consequently, the loss of astrocytes in NMO-associated optic neuritis may impair remyelination, an adaptive response to injury with trophic, protective, ion channel and energetic ramifications.
The depletion of astrocytes within the proximity of active CNS injury in NMO may also result in a protracted state of demyelination resulting in an inefficient method of axonal transmission by membrane rather than saltatory (nodal/paranodal) conduction. In the healthy CNS, astrocytic processes drape themselves around the axons at myelin internodes and serve almost as a ‘place-setting’ for the orderly positioning of myelin wraps. When astrocyte loss is coupled with chronic demyelination, the intrinsic organisation of myelination and the clustering sodium channels at the nodes of Ranvier and paranodal regions is altered, and axonal conduction becomes inefficient. Interestingly, mice lacking glial fibrillary acidic protein (GFAP), an astrocyte-specific intermediate filament, demonstrate impaired optic nerve myelination, implicating the importance of astrocyte integrity in myelination.58
The much broader distribution of sodium channels necessary to reconstitute axonal conduction in the setting of chronic demyelination is commensurately associated with a greater energetic demand on intra-axonal mitochondria.59 The greater demand for ATP is at least partly related to the need to establish and maintain an altered equilibrium, secondary to multiple derangements in ion currents and altered channel characteristics. For example, newly synthesised sodium channels undergo pore closure, and thereby cessation of membrane depolarisation, in response to elevated temperature resulting in the clinical manifestation of Uhthoff's phenomenon. In addition, the accumulation of intra-axonal sodium and calcium can result in mitochondrial damage and bioenergetic failure through the production of reactive oxygen species. Potential players include inactivating sodium channels (eg, Nav1.6), sodium/calcium exchangers (eg, NCX), glutamate receptors (eg, GluR), acid-sensing channels (eg, ASIC1), cation channels (eg, TRPM4) and voltage-dependent calcium channels (eg, VDCC).12 ,60 The resulting intra-axonal energetic supply-demand mismatch can eventually provoke the liberation of excitatory amino acids, such as glutamate, culminating in irreversible axonal injury and neuronal demise, the presumed underpinning of chronic disability in AON. In support of this mechanism, a small trial using the N-methyl-d-aspartate receptor antagonist, memantine, demonstrated less thinning of the RNFL in treated versus placebo participants.61 Currently, the sodium channel-blocking agent, phenytoin, and the acid-sensing channel blocker, amiloride, are being used in clinical trials in AON. These clinical trials use RNFL thickness, as measured by OCT and SLP, as surrogate measures of neuroprotection.
Recent work has identified a small group of patients who present with AON but whose clinical course demonstrates atypical recurrent activity or dependency on immunosuppression. In 2003, Kidd et al62 described a cohort of patients who presented with painful, subacute optic neuropathy and responded promptly to treatment with systemic corticosteroids. These individuals relapsed rapidly on steroid withdrawal, and the condition was labelled chronic relapsing inflammatory optic neuropathy (CRION) to distinguish it from typical optic neuritis. Arndt et al63 described an additional cohort of patients with recurrent isolated optic neuritis (RION) who experienced repeated attacks of AON that resolved without treatment but resulted in progressive vision loss over time. No patients with CRION or RION presented with or developed MRI lesions consistent with MS, and testing for antibodies against AQP-4 revealed that only a small fraction of these individuals suffered from NMO.64 Patients with CRION or RION likely represent a small fraction of AON with a unique immunopathology. This could be due to targeting of a unique optic nerve-specific antigen or a deficient T regulatory response, or perhaps it is a consequence of a novel inflammatory response. The prospective evaluation of patients with acute CRION and RION with immunological measures, OCT and electrophysiology will be critical for identifying novel diagnostic and therapeutic approaches.
New therapies for the treatment of acute demyelinating injury
The array of MS therapies has increased rapidly over the past decade. Therapies approved in the 1990s, β-interferon and glatiramer acetate, have been joined by new treatments with novel mechanisms of action: inhibition of leucocyte adhesion (natalizumab); interference with S1P-mediated lymph node egress (fingolimod); interference with lymphocyte replication (teriflunomide) and activation of the oxidative stress response pathway (dimethyl fumarate). The ability of these therapies to minimise injury or promote recovery after acute demyelination, however, has rarely been evaluated. Based on their rapid onset and CNS penetration, some of these newer MS therapies may offer promise in limiting vision loss and facilitating recovery after AON. A single dose of natalizumab, administered soon after the onset of an MS relapse, did not hasten clinical recovery but decreased Gd-enhancing lesion volume.65 The lack of a demonstrable change in the rate of clinical recovery may have been due to the variety of clinical presentations, the insensitivity of the clinical measure (Expanded Disability Status Scale) and the lack of sensitive paraclinical tools to assess the impact on anatomic and physiological changes. Indeed, fingolimod recently has shown efficacy in ameliorating AON in the experimental autoimmune optic neuritis animal model when administered during the effector phase of the disease.66 A similar effect has been noted using dimethyl fumarate after the onset of CNS inflammation in the experimental autoimmune encephalomyelitis model.67 The potential neuroprotective action of fingolimod and dimethyl fumarate in these animal models suggests that these agents may be ideally suited for the acute treatment of AON.
There are now therapeutic approaches with the capability of inducing remyelination in experimental animal models. In a toxin-induced rodent model of demyelination, an antibody against LINGO1, a CNS protein that acts as a negative regulator of oligodendrocyte precursor differentiation, promoted CNS remyelination by creating a microenvironment conducive to oligodendrocyte differentiation.68 Anti-LINGO1 monoclonal antibody has been humanised and is being tested in early clinical trials of patients with MS. Reparative agents such as anti-LINGO1 may offer a unique avenue for facilitating the restoration of visual function in AON.
Advancements in the understanding of the mechanisms underlying acute CNS demyelination, novel developments in the ability to measure axonal and myelin injury, treatments with novel anti-inflammatory and immunomodulatory mechanisms, and an unmet therapeutic need warrant a renewed investigation of the treatment of AON. Clinical research on AON with both established and new agents can provide information on multiple complementary outcomes: clinical recovery, tissue preservation and remyelination. The resulting data on visual acuity (high and low contrast), visual fields, colour vision, peripapillary RNFL thickness, GCL+IPL thickness, ONHC responses in mfERG, mfVEP and DTI will provide an elaborate framework in which to decipher a specific agent's effect on inflammation, axonal integrity, neuronal survival, oligodendrocyte injury and remyelination.
Clinical trials for AON therapy
Given the significant visual improvement (median recovery 20/16) observed in the ONTT placebo group,5 future clinical trials to evaluate new AON treatments will likely require significant patient numbers to ensure adequate powering. As a result, the need for substantial patient enrolment may dissuade the assessment of certain compounds with potentially moderate clinical effects. Therefore, the analysis of retinal architecture by OCT and related methods may prove to be a valuable surrogate for neuroprotection after acute inflammatory injury. Indeed, Henderson et al27 have estimated sample sizes for clinical trials of therapeutic agents in AON, when using OCT as the outcome measure, and demonstrated that as few as 75 patients per treatment arm are needed to provide 90% power for a modest 40% effect size. The increased sensitivity of GCL+IPL measurement may reduce enrolment requirements further.22 A positive result from a smaller OCT trial may provide the needed impetus to investigate the impact on clinical outcomes in a larger phase 3 trial.
Future directions
Advances in our understanding of demyelinating injury and the development of novel structural and physiological metrics of optic nerve integrity provide a promising environment for future translational and clinical research in AON (figure 1). In addition, the close relationship between idiopathic AON and other CNS demyelinating lesions allows the results of AON studies to immediately impact our understanding of MS pathophysiology and treatment. Using sensitive metrics such as OCT, ONHC, mfVEPs and DTI, future investigations may simultaneously address questions regarding the mechanism and timing of oligodendrocyte and RGC injury, the beneficial effects of immunomodulatory and neuroprotective therapies, and the efficacy of restorative interventions.

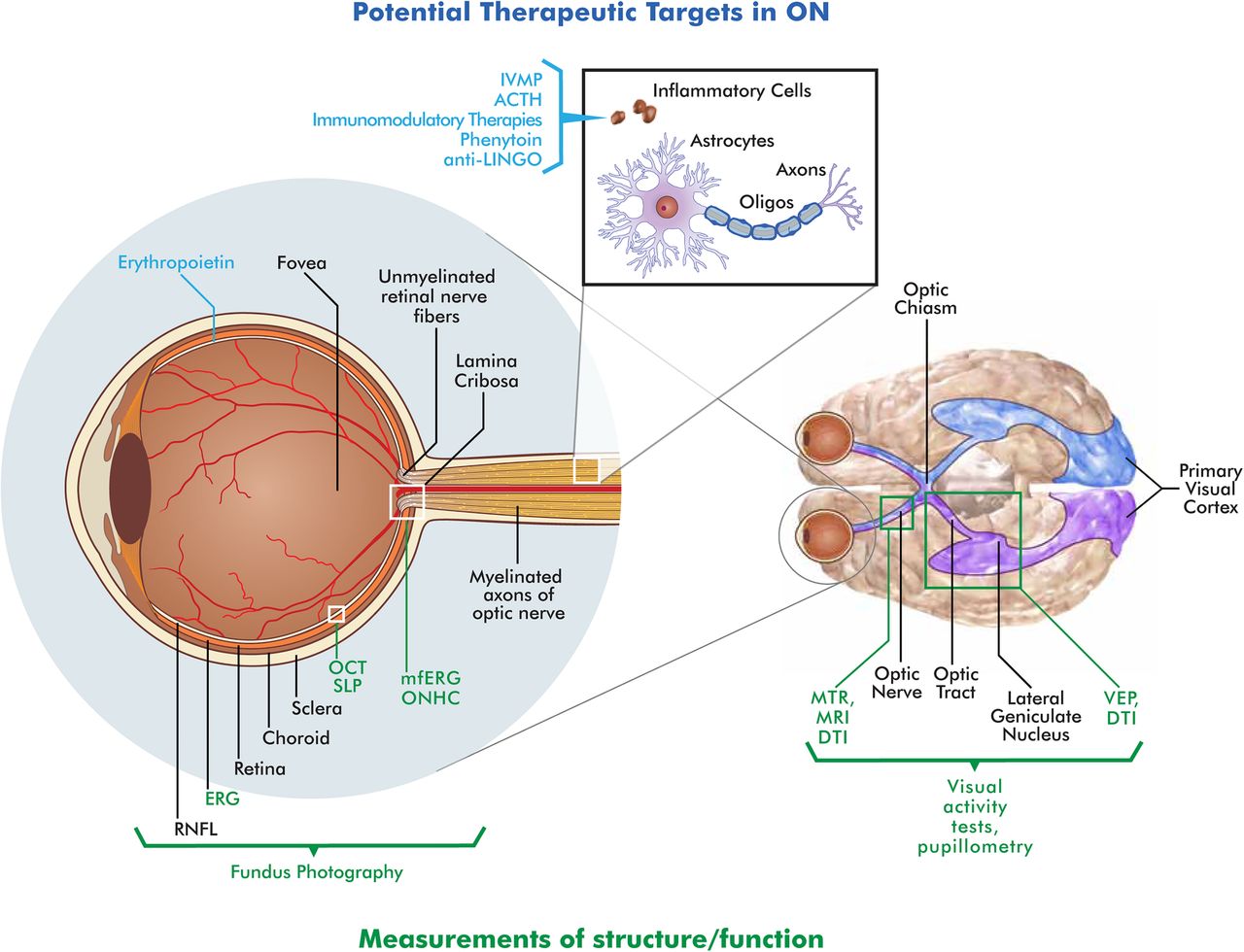{kind=link}
Schematic of the retina, optic nerve and postchiasmal afferent visual system. Potential therapeutic targets (blue text) and measures of visual function (green text) are illustrated above and below the diagram, respectively (RNFL, retinal nerve fibre layer; ERG, electroretinogram; OCT, optical coherence tomography; ONHC, optic nerve head component; SLP, scanning laster polarimetry; VEP, visual evoked potential; DTI, diffusion tensor imaging; MTR, magnetisation transfer imaging ratios).
With these objectives in mind, a future AON treatment trial need only utilise a few sites to enrol a sufficient number of participants to evaluate multiple effects of a therapeutic agent. For example, a recent two-institution clinical trial (clinicaltrials.gov; NCT01838174) has been designed to compare the anti-inflammatory, neuroprotective and restorative effects of ACTH and methylprednisolone using OCT and electrophysiology. Using OCT of the peripapillary RNFL and macula, the trial will compare RNFL loss and GCL+IPL thinning to compare the neuroprotective effects of these compounds on axonal and RGC survival. In addition, OCT will be used to examine whether the differential anti-inflammatory effects of ACTH and methylprednisolone affect the rate of resolution of optic nerve head oedema and the timing of RNFL thinning and RGC loss. Sensitive electrophysiological metrics such as mfVEPs and the ONHC of the mfERG will be used in concert with OCT to evaluate the extent of and recovery from demyelinating injury.
While novel in integrating many new approaches for the evaluation of optic nerve integrity and function, this study highlights many of the unresolved questions facing the optimal design of clinical trials in AON. What is the optimal window for the enrolment and institution of therapy in AON? Small trials with encouraging results on neuroprotection suggest the window may be small (8–10 days). Ultimately, however, the therapeutic window may vary based on the mechanism of action of the agent (ie, anti-inflammatory or neuroprotective), method of administration (oral, intravenous, retrobulbar) and pharmacodynamics (time to therapeutic level/effect in target tissue). What is the optimal outcome measure? Is it anatomic (RNFL thickness), electrophysiological (mfVEP, ONHC) or functional (VF, LCVA)? As noted previously, each metric has merits and limitations. In the short term, a pragmatic approach may be best. Sensitive measures of axonal, glial or neuronal injury such as OCT and mfVEP may be ideal to identify treatment effects in small cohorts. Afterwards, compounds with significant results can be moved forward into larger trials designed to document clinical improvement in AON and other demyelinating injuries. The data acquired from this and other ongoing AON trials will likely provide the foundation for using AON as a paradigm disorder for clinical and translational studies of demyelinating disease.
Acknowledgments
The authors thank MedVal Scientific Information Services, LLC, for editorial assistance.
References
Footnotes
-
Contributors JLB, EMF – conceived, designed and drafted the review. MN, FC, RCS, JCC, SLG, LJB, CEM, TV, MM, MLM, AWT, TWP, TF – performed critical revisions of the manuscript for important intellectual content.
-
Funding Editorial assistance was funded by Questcor Pharmaceuticals, Inc.
-
Competing interests All authors attended a scientific advisory board hosted by Questcor Pharmaceuticals, Inc. JLB receives grants from the Guthy Jackson Research Foundation and the NIH (EY022936); serves as a consultant for Novartis Pharmaceuticals, Alnaylam Pharmaceuticals, MedImmune, Chugai Pharmaceuticals, EMD Serono, Abbott Pharmaceuticals, Genentech, Genzyme and Questcor Pharmaceuticals; receives licence royalties for a patent re Compositions and Methods for the Treatment of Neuromyelitis Optica; and serves on the editorial boards of the Multiple Sclerosis Journal and Journal of Neuro-ophthalmology. MN employee of Questcor Pharmaceuticals, Inc. FC receives grants from the Multiple Sclerosis Society of Canada, speaker fees from EMD Serono and serves as a consultant for Novartis Pharmaceuticals. RCS serves as a consultant for Biogen-Idec, EMD Serono, Teva Neuroscience, Lundbeck, Pfizer, Thrombogenics, Sanofi-Aventis, Novartis Pharmaceuticals, BioClinica, Covance, Glaxo-Smith-Kline, Questcor, Heidelberg Engineering, Merck, United States Department of Defense, and the Food And Drug Administration; receives speaker honoraria from Biogen-Idec, EMD Serono, Teva Neuroscience, Lundbeck, Pfizer, Novartis Pharmaceuticals, Sanofi-Aventis and Questcor. JCC receives research support from Novartis Pharmaceuticals, Biogen-Idec and Roche; receives speaking fees from Acorda Therapeutics, Novartis Pharmaceuticals, Biogen-Idec, EMD Serono, Genzyme, Bayer Healthcare, Acorda Therapeutics, Questcor and Teva Neuroscience; and receives consultant fees from Novartis Therapeutics, Biogen-Idec, Genzyme, Acorda Therapeutics and Questcor. SLG serves as a consultant for Vaccinex and Biogen-Idec. LJB receives consulting fees from Biogen-Idec, Questcor and Vaccinex. CEM serves as a consultant for Biogen-Idec, Teva Neuroscience, Bayer Healthcare, EMD-Serono, Questcor, Novartis, Genzyme, Roche and Genentech. TV receives speaker honoraria and consulting fees from Biogen, Teva Neuroscience, Novartis, Genzyme, EMD Serono and Questcor. MM receives research support from Novartis Pharmaceuticals. MLM receives speaker honoraria from Biogen-Idec and Novartis Pharmaceuticals and research support from Acorda Therapeutics. AWT receives consulting fees from Palatin Technologies. TF receives speaker honoraria and consulting fees from Biogen-Idec, Teva Neuroscience, Novartis Pharmaceuticals, Genzyme and Acorda Therapeutics. EMF receives speaker honoraria and consulting fees from Biogen-Idec, Teva Neuroscience, Novartis Pharmaceuticals, Genzyme and Acorda Therapeutics.
-
Provenance and peer review Not commissioned; externally peer reviewed.