Article Text

Abstract
Background Amyotrophic lateral sclerosis (ALS) is a progressive and usually fatal neurodegenerative disease. Survival from diagnosis varies considerably. Several prognostic factors are known, including site of onset (bulbar or limb), age at symptom onset, delay from onset to diagnosis and the use of riluzole and non-invasive ventilation (NIV). Clinicians and patients would benefit from a practical way of using these factors to provide an individualised prognosis.
Methods 575 consecutive patients with incident ALS from a population-based registry in South-East England register for ALS (SEALS) were studied. Their survival was modelled as a two-step process: the time from diagnosis to respiratory muscle involvement, followed by the time from respiratory involvement to death. The effects of predictor variables were assessed separately for each time interval.
Findings Younger age at symptom onset, longer delay from onset to diagnosis and riluzole use were associated with slower progression to respiratory involvement, and NIV use was associated with lower mortality after respiratory involvement, each with a clinically significant effect size. Riluzole may have a greater effect in younger patients and those with longer delay to diagnosis. A patient's survival time has a roughly 50% chance of falling between half and twice the predicted median.
Interpretation A simple and clinically applicable graphical method of predicting an individual patient's survival from diagnosis is presented. The model should be validated in an independent cohort, and extended to include other important prognostic factors.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS, also known as motor neuron disease) is a degenerative disease of the nervous system which is progressive, incurable and almost always fatal. Around 1 in 400 people will develop the condition, usually aged between 50 and 70 years.1 The cause is not well understood; there is a significant genetic component, but only a minority of cases are due to single-gene mutations.2
The rate at which the condition progresses varies greatly, as does the length of time from symptom onset to death, from a few months to more than 10 years.3 It is therefore difficult to advise a patient on how long they may expect to live, and on the uncertainty in this prediction, and not every patient wants detailed information; nonetheless, for many patients diagnosed with a life-limiting illness, such an estimate is important for providing hope and enabling them to plan their life and its ending, not to mention improving the design and interpretation of clinical trials. A recent systematic review3 and more recent studies4–9 have found evidence that a number of factors can help to predict longer survival in people with ALS. These include younger age at symptom onset, longer delay from symptom onset to diagnosis (diagnostic delay), limb onset as opposed to bulbar onset, lack of cognitive impairment, lack of respiratory muscle weakness, certain genetic factors, and the baseline and slope of certain laboratory measures and severity scores. The evidence that gender and upper-limb onset (as opposed to lower limb) can independently contribute to predicting survival is conflicting.
Three medical treatments have also been shown to prolong survival: non-invasive ventilation (NIV) when respiratory support is necessary, gastrostomy feeding when dysphagia is present, and riluzole. As a drug treatment which has been shown to prolong survival in a neurodegenerative disease, riluzole is of great theoretical as well as practical interest, but it is not known whether the survival improvement occurs throughout the disease course or only at certain stages.
A practical way of combining the factors to produce a good prediction for a particular individual would be of great use to the clinician and the affected person. While there are many multivariate survival analyses in the literature, most would require the clinician to reconstruct the mathematical model (and in many cases the necessary information is not given). Further, most use a Cox's proportional hazards model; in a similar study of mortality in ALS,10 the use of such a model was examined and found to be ‘clearly inadequate’, as the effects of predictors including riluzole use were found to vary with time from diagnosis, violating the proportional hazards assumption. We know of only two explicit predictive models, of which one11 requires data which would not usually be available in the clinic, and the other12 omits important predictors.
To develop such a model, one would need a large population-based cohort of patients with newly diagnosed ALS, with consistently recorded data on the known prognostic factors and length of survival, along with a robust statistical technique requiring a minimum of arbitrary assumptions and informed by understanding of the biology of the disease. The South-East England register for ALS (SEALS) registry provides such a data set, ascertaining all cases of ALS in a population of some 3 million in the South-East of England. Full details of the registry and case ascertainment are available elsewhere.13
The dominant cause of death in ALS is respiratory muscle weakness, causing respiratory failure or infection: estimates range from 65% to 89%.14–19 The true proportion is probably higher, as several studies investigating this included categories such as ‘unexplained sudden death’ which could well have a respiratory cause. As a simplifying assumption, we therefore modelled the course of ALS in terms of four consecutive events: symptom onset, diagnosis, respiratory involvement and death; our goal was to generate a clinically useful model, able to inform clinicians and patients about survival.
Methods
Cases and variables
The criteria for inclusion in the SEALS registry are described elsewhere,13 but in brief, cases were included if they presented between January 1990 and May 2013, and received a diagnosis of ‘definite’ or ‘probable’ ALS according to the El Escorial criteria.20 A few patients could not be included because they experienced onset at two sites simultaneously, had respiratory muscle weakness at onset or only received a diagnosis postmortem.
We included in the model the most significant and easily assessed factors that have been found to be associated with survival, namely site of onset (bulbar, upper limb or lower limb), age at symptom onset, gender, time from onset to diagnosis (diagnostic delay) and treatment with riluzole. Riluzole use was analysed as a binary variable: if a patient had ever taken riluzole, it was coded as 1, otherwise 0. This method gives the closest result parallel with the intention-to-treat method of analysis used in clinical trials. Insufficient information was available on cognitive impairment, respiratory muscle weakness, genetic status, use of NIV and gastrostomy feeding. Analyses relating to survival in patients receiving NIV were performed using data from patients attending the Motor Nerve Clinic at King's College Hospital, London (table 1), referred to below as the ‘NIV dataset’.
NIV data set: descriptive statistics
Summary of the statistical model
The technical details of the model are given below, but the reasoning behind it can be summarised as follows. We observed that patients with ALS using NIV have a constant risk of death, namely 20% per month in bulbar-onset cases and 10% per month with limb onset; this does not change significantly over time. Noting that most deaths in ALS occur in the context of respiratory muscle weakness, we inferred that this death rate would apply from the onset of respiratory muscle involvement, and that the rate would also be constant (albeit higher) in patients with respiratory muscle weakness who were not using NIV—we used data from the largest suitable study21 to estimate how much higher.
Most patients do not have respiratory muscle involvement at diagnosis, but develop it later on after a variable length of time. We found that it fitted the observed survival data very well if we assumed that the logarithm of the time from diagnosis to respiratory muscle involvement was normally distributed. Using this model, we could ask whether the predictor variables (age at symptom onset, diagnostic delay, riluzole use) were associated with progression to respiratory muscle weakness, or separately with mortality after respiratory muscle involvement.
Mortality after respiratory involvement
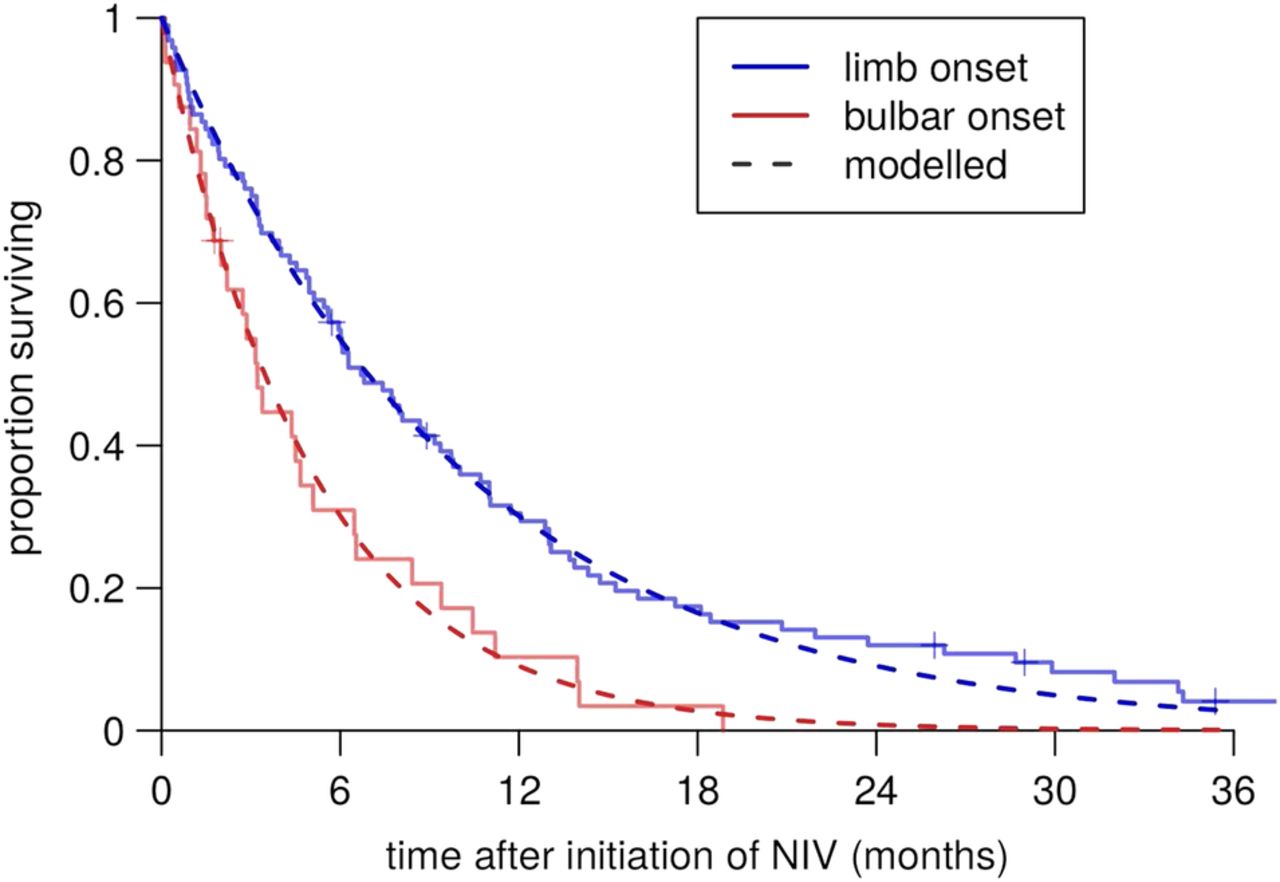
Our data did not include the date of onset of respiratory involvement. We therefore examined Kaplan-Meier plots of the NIV data set, which suggested that mortality in patients on NIV (all of whom have respiratory involvement) is well modelled by an exponential distribution, that is, a constant mortality rate, at least until survival falls below 20% (figure 1). Extrapolating this back, we treated mortality after respiratory involvement as constant, and estimated the rate at 20% per month in bulbar-onset cases and 10% per month in limb-onset cases, for patients using NIV. We examined the prognostic factors for their association with mortality in this data set using univariate exponential regression.

Constant-hazard fitted post-non-invasive-ventilation (post-NIV) survival curves in the clinic data set.
For patients with respiratory involvement but not using NIV, these mortality rates needed to be adjusted for the survival benefit of NIV. We were not able to estimate this survival benefit using our data, and estimates in the literature vary widely. The largest study is reported by Bourke et al,21 but the authors do not explicitly derive a figure for survival benefit. Fitting exponential distributions to their Kaplan-Meier plots suggests a 3.5-fold survival benefit for NIV (figure 2). On the basis of the clinic audit data, we estimated that 80% of our patients accessed NIV during their illness, and assumed this to be independent of the covariates.

Reanalysis of data from Bourke et al.21 NIV, non-invasive ventilation.
The combined model
Between the four modelled events (symptom onset, diagnosis, respiratory involvement and death) lie three time intervals, which were modelled separately. As our goal was to create a clinically useful model, we chose to model survival from diagnosis, as it would be at least as useful as predicting from onset and would allow us to use the time from symptom onset to diagnosis (diagnostic delay) as a predictor variable.
The effects of predictor variables on survival can be modelled in two ways: as affecting the chance of death occurring at any particular time (a Cox's proportional hazards model) or as affecting the length of time until death (an accelerated failure time (AFT) model). In the case of time from diagnosis to respiratory involvement, we were dealing with the progression of a condition rather than the stochastic occurrence of an event, and so an AFT model was appropriate.
To maximise the statistical power, we chose to use a parametric model with an a priori choice of modelling distribution. The distribution of times from diagnosis to respiratory involvement could not be visualised directly, but only in the context of the combined model, so the choice of model distribution was arbitrary among those amenable to AFT analysis. A Weibull distribution was tried first, resulting in a poor fit; a log-normal distribution gave a much better fit. To avoid overfitting, the log-normal distribution was chosen without trying other distributions.
The overall survival time distribution was derived as the convolution of the log-normal distribution of time from diagnosis to respiratory involvement with the cumulative exponential distribution of time from respiratory involvement to death.
Fitting the model
The combined model was then fitted to the survival data. Two parameters were allowed to vary in fitting the model: the log-scale mean of the log-normal distribution of time from diagnosis to respiratory involvement, and its SD. The mean was the only parameter which was allowed to depend on the predictor variables of age at symptom onset, diagnostic delay (log transformed) and riluzole use. The model was fitted by maximum-likelihood estimation, using the flexsurv package in R (C Jackson. Flexsurv: Flexible parametric survival models. R package version 0.2. 2013. http://cran.r-project.org/package=flexsurv).
Fitting the model to each group of patients according to onset site (bulbar, upper limb or lower limb) gave substantially closer fits than the data set as a whole. This was due to a greater spread (log-scale SD) of the log-normal distribution in the limb-onset groups than in the bulbar-onset group. The two limb-onset groups were practically identical in all analyses, so we pooled the limb-onset groups but analysed the bulbar-onset group separately.
All analyses were conducted and figures produced using R 2.15.2 (R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2012. ISBN 3-900051-07-0. http://www.r-project.org/) running on Linux (Lubuntu 13.04).
Results
Descriptive statistics and model fit
A total of 575 patients were included. Very few cases were excluded due to missing dates of birth, diagnosis or onset or missing data on predictor variables; patients who remained alive at the time of analysis were included, treating survival as censored, but in fact the date of death was known in 97% of cases (table 2), so there was no risk of bias due to informative censoring.
SEALS data set: descriptive statistics
Table 2 shows summary statistics of the variables of interest in the patient groups. These were comparable to cohorts from other centres; for example, the median age at symptom onset was 61.5 and the mean 60.7, which fall close to the average for population-based cohorts.3 Table 3 shows the strengths of the associations between them. In particular, there were mutual associations between older age at symptom onset, bulbar onset and female gender. Figure 3 shows Kaplan-Meier plots of survival time from diagnosis for the bulbar-onset and limb-onset groups. In figure 4, the modelled survival curves are superimposed on these plots, suggesting an excellent fit of the model to the data.
Pairwise associations between the variables
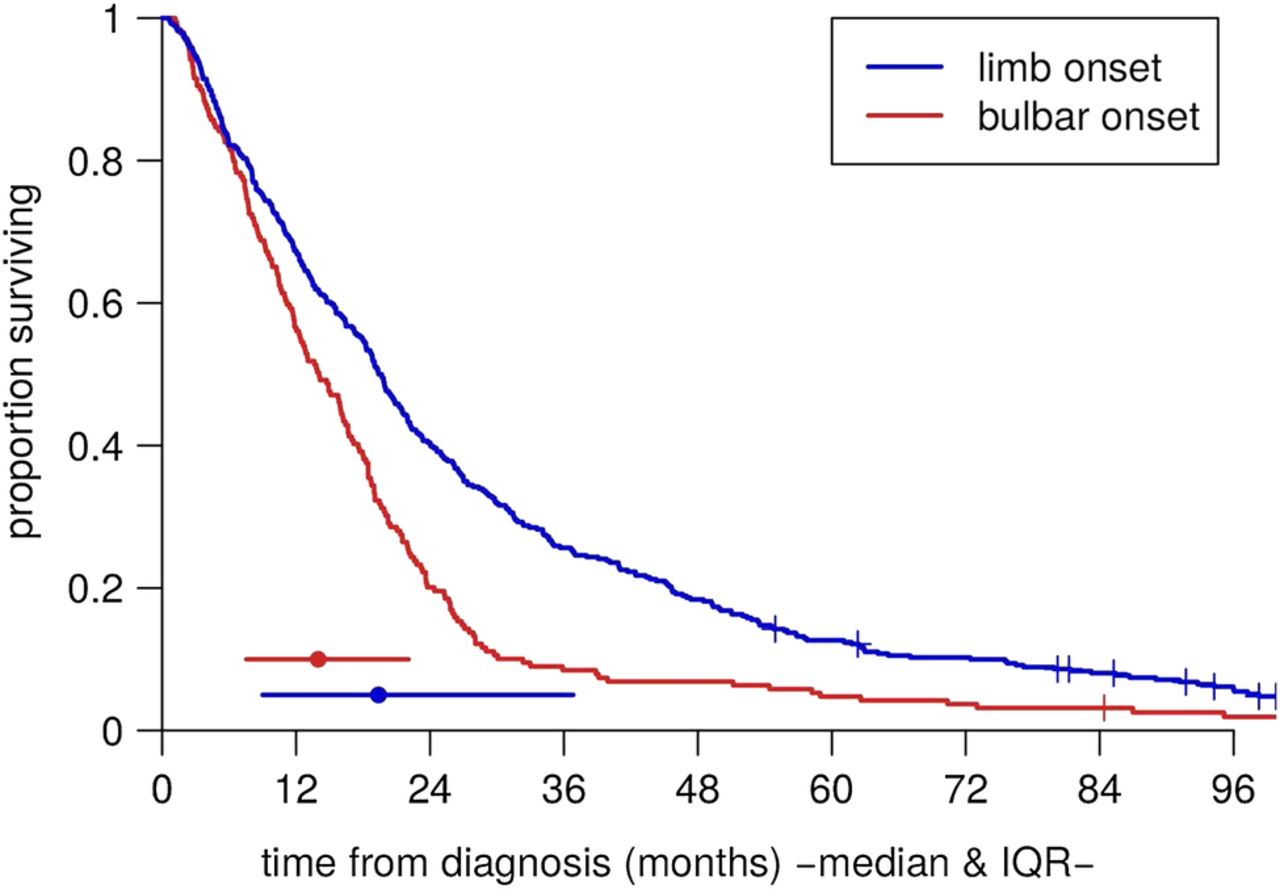
Raw Kaplan-Meier survival curves for the bulbar-onset and limb-onset groups.

Fitted survival curves.
Mortality in patients on NIV
No significant associations were found between mortality in the NIV data set and age at symptom onset, diagnostic delay, gender or riluzole use (table 4), and we therefore did not include these variables in the mortality part of the combined model. However, because of the relatively small sample size, the CIs are large, and the analysis does not rule out the possibility of clinically important effects.
Effects of covariates on survival in patients on NIV: univariate analyses
Progression to respiratory involvement
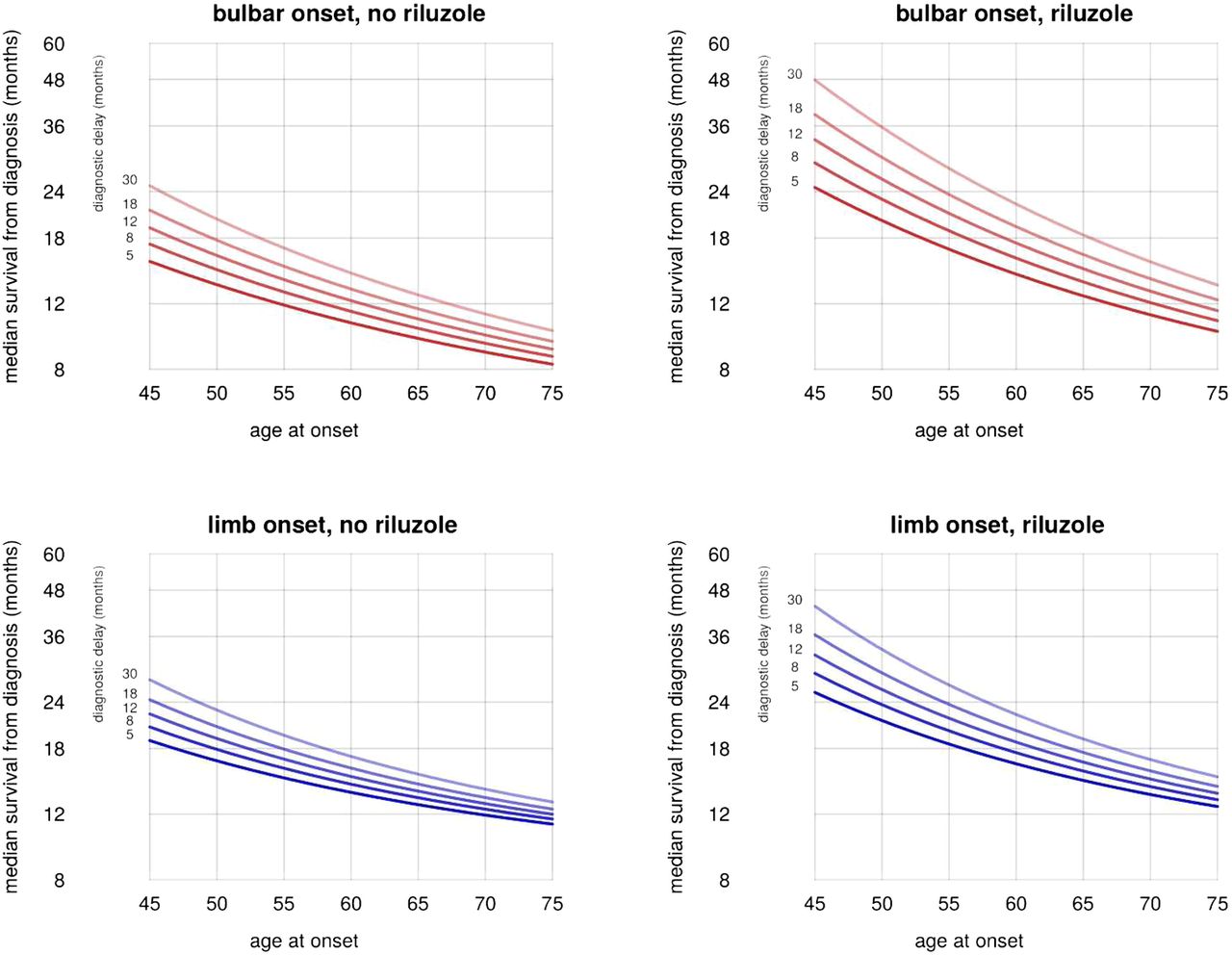
Age, diagnostic delay and riluzole use were each independently associated with slower progression from diagnosis to respiratory involvement. This held true in univariate (table 5) and multivariate (table 6) analyses, for bulbar-onset and limb-onset cases separately, and the effect sizes were all clinically relevant (figure 5). No significant effect was found for gender.
Effects of covariates on time from diagnosis to respiratory involvement: univariate analyses
Effects of covariates on time from diagnosis to respiratory involvement: multivariate analysis

Effects of predictors on time to respiratory involvement.
Riluzole appears to have a greater benefit, even in ratio terms, in younger patients and in those with longer diagnostic delay (ie, in patients with longer predicted survival; figure 5). In an unplanned post hoc analysis, statistically significant interaction effects were found in the limb-onset group but not the bulbar-onset group, although the effect size estimates were slightly larger in the bulbar-onset group.
Using the graphs to predict survival in clinic
To predict the survival time of a patient at diagnosis, we suggest the following procedure. If the patient already has respiratory involvement, their mortality is given simply by 20% per month if there is bulbar involvement, or 10% per month if there is not; if the patient is not using NIV, these figures increase to 54% and 31%, respectively. For patients without respiratory involvement, figure 5 gives the predicted median survival time, which can be used to estimate 50% CI for the estimate: the survival time for half of cases will fall between half the median and twice the median.
For example, if figure 5 suggests a median survival of 12 months from diagnosis for a particular patient, there is a 50% chance that they will survive for between 6 and 24 months. At the time of diagnosis, riluzole use will not usually be known: the model allows prediction with and without riluzole, giving the clinician and patient a useful estimate of the effect of riluzole in the individual case.
Eighty per cent of our cases had an age at symptom onset between 45 and 75, and 80% had a diagnostic delay between 5 and 30 months. The model should be interpreted with caution for cases lying outside these ranges.
Testing for robustness
Although AFT models are considered relatively robust in general, we wished to test our model in detail.
To examine the sensitivity of the model to changes in the data, the analyses were rerun many times, removing 20% of cases at random each time. For post-NIV mortality, 95% of the 500 resulting estimates lay between 17.1% and 24.4% (bulbar onset) or 8.6% and 10.8% (limb onset) monthly mortality. The results for the combined model are shown in table 6; in general, more than 95% of the estimates lie within the 95% CI of the primary analysis. It is also strongly reassuring that the estimated effect size of each predictor variable was practically the same in the bulbar-onset and limb-onset groups.
The figures for the survival benefit of NIV (3.5×), and for the proportion of our cases who received NIV (80%), were derived from sources external to the study. Varying these figures within ranges of 2× to 6× and 50–90%, respectively, the coefficients for age at symptom onset, diagnostic delay and riluzole all remained within 22% of the values from the main analysis.
The choice of a log-normal distribution to model time from diagnosis to respiratory involvement was arbitrary. However, the fit remained good when the data were stratified by the predictor variables, and the distribution also fitted well to the data derived from the patients attending the Motor Nerve Clinic at King's College Hospital, London (of which the NIV data set used in the present study was a subset; figure 6). This cohort is not suitable for validating the model as a whole, since it is not population based and partially overlaps with the SEALS cohort, but it helps to confirm that the shape of the chosen distribution was appropriate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Survival curves fitted to the KCH Motor Nerve Clinic data set.
Discussion
Findings
We present a straightforward graphical method of predicting a patient's survival time from ALS diagnosis, taking into account the known prognostic factors of onset site, age at symptom onset, diagnostic delay, riluzole use, NIV use and respiratory muscle involvement. The model may predict a median survival of as much as 4 years or as little as 8 months, and even less in patients with respiratory involvement. The true survival time falls between half and twice the median in about half of the patients. Pending validation of the model, we propose this as a clinically useful tool.
Comparison with existing models
The present study improves on the existing prognostic models in several important ways. We give an explicit, straightforward method which can be applied in the clinic in moments, without specialised tests or detailed calculation. It provides an individualised estimate, not merely a broad prognostic category, along with a guide to the uncertainty in this estimate which is easy to calculate and interpret. Using a large population-based cohort avoids biases associated with referral to and treatment in a specialist clinic; we avoid making the proportional hazards assumption, found to be invalid in a similar study;10 and our main analysis is multivariate, taking into account all predictor variables at the same time. Finally, and crucially, it can readily be validated by other investigators in independent cohorts of patients.
Riluzole use was associated with slower progression to respiratory involvement in this study. Riluzole is known to prolong overall survival in ALS, but its effects on particular stages of the condition have not previously been reported. While our data do not rule out a continuing effect of riluzole on mortality after respiratory involvement, this finding emphasises the importance of starting riluzole treatment as soon as possible after diagnosis. Our analysis also suggests that riluzole may be especially beneficial for younger patients and those with longer diagnostic delay.
The association of bulbar onset with shorter median survival is accounted for by higher mortality after respiratory involvement, a higher average age at symptom onset and less use of riluzole in the bulbar-onset group. Patients with bulbar weakness are at higher risk of aspiration and respiratory tract infection, a common mode of death; they are also less likely to tolerate NIV. While the present analysis related to bulbar involvement at onset, it is clinically plausible that the effect on mortality would be seen in any patients who have bulbar weakness at the time of respiratory involvement. We have not found evidence that patients with bulbar-onset progress to respiratory involvement sooner, after allowing for age and riluzole use.
The effects of known prognostic factors were confirmed in our model, with similar effect sizes: for example, a recent study22 found a 26% reduction in hazard for riluzole, which is consistent with figure 5. Since our statistical methods have not been used previously in this context, this provides a check on our methods and indirectly supports the novel findings of this study.
Limitations
Like any model, the present study represents a compromise between simplicity and robustness on the one hand, and the faithful representation of reality on the other.
There is evidence that patients with upper motor neuron features alone12 or lower motor neuron features alone12 ,22–24 tend to survive longer, and that those with cognitive impairment,25 neck weakness,26 weight loss at diagnosis6 and certain genotypes27 ,28 have a higher overall mortality. Our study did not include information on these factors.
In constructing the model, we assumed that the patients did not have respiratory muscle involvement at diagnosis. This biases our model slightly towards predicting shorter survival in those without respiratory involvement. The basic assumption that respiratory involvement always occurs before death will also have exceptions. The availability and use of gastrostomy and NIV may vary between centres, as well as other aspects of care relevant to survival.29 The model does not take account of such variation. Although we tried to avoid bias by using a population-based cohort, one must always be cautious in applying a model to populations other than the one from which it was derived.
The model is useful for predicting survival at the time of diagnosis, and once respiratory involvement has occurred; the prediction of survival at other times during a patient's illness is not addressed by the model.
The present study is not a clinical trial, and therefore the effect of riluzole use should be interpreted cautiously as ‘those who receive riluzole survive longer’ rather than as ‘riluzole prolongs survival’, though both are consistent with the data.
A weakness of this study is that the population studied has a lower median age of onset than is typical for population registers. This may reflect underascertainment of older age groups, which would be consistent with the lifetime risk curves previously reported.1 ,30 It may alternatively reflect the influence of London, which is a large component of the catchment and being urban, has a younger population than average. A further weakness is that the risk curves have been derived in a population study but will be most likely applied in a clinic setting, and clinics have a generally younger, more male population, which may limit its applicability.
Future work
Our model makes quantitative statements about the effects of prognostic factors on the distribution of survival time in ALS, and suggests the specific testable hypotheses that riluzole treatment may be more beneficial in patients who do not yet have respiratory involvement, in younger patients and in those with longer diagnostic delay. Analysis of an independent population-based cohort is necessary to investigate these findings and predictions.
Instead of using mortality data, the speed of progression of ALS can also be assessed using the time from clinical onset to the time of involvement of the second site. We would predict that age, diagnostic delay and riluzole use would be associated with this, but that gender and site of onset would not have clinically significant independent effects.
References
Footnotes
Contributors JAK and AA-C were involved in the conception, design of the study and data interpretation. NK, AK, PNL, SM, CES, MT, AA-C contributed to the acquisition of the data. JAK analysed the data. All authors were involved in critically revising the manuscript and approving the final version to be submitted, and agreed to be accountable for all aspects of the work. The corresponding author had full access to all the data in the study and has final responsibility for the decision to submit for publication.
Funding This work was funded by the UK Medical Research Council and Economic and Social Research Council under the aegis of the EU Joint Programme—Neurodegenerative Disease research project. AAC and CES receive salary support from the National Institute for Health Research (NIHR) Biomedical Research Centre and Dementia Biomedical Research Unit at South London and Maudsley NHS Trust and King's College London. The work leading up to this publication was funded by the European Community's Health Seventh Framework Programme (FP7/2007–2013; grant agreement number 259867). The funding sources had no role in writing the manuscript or deciding to submit it for publication.
Competing interests PNL reports grants from MND Association Funding to support MND Clinic from 1995 to 2010, during the conduct of the study. CES reports consultancy and/or collaborative relationships with Glaxo Smith Kline, Vertex Pharmaceuticals, Chronos Therapeutics and Eli Lilly; he receives no personal gain and has never had any shares of any sort. AAC reports grants from EU FP7, grants from EU JPND—MRC and ESRC, grants from NIHR BRC in Mental Health and Dementia BRU, during the conduct of the study. All other authors declare no conflicts of interest.
Ethics approval The study was approved by the local ethics committees of King's College Hospital (15/LO/0810) and South London and Maudsley (222/02) NHS Foundation Trusts.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary