Article Text

Abstract
Objectives To examine the influence of the glucocerebrosidase (GBA) mutation carrier state on age at onset of Parkinson’s disease (PD), the motor phenotype and cognitive function at baseline assessment in a large cohort of UK patients. We also analysed the prevalence of mood and behavioural problems that may confound the assessment of cognitive function.
Methods We prospectively recruited patients with PD in the Tracking Parkinson’s study. We fully sequenced the GBA gene in all recently diagnosed patients (≤3.5 years). We examined cognitive (Montreal Cognitive Assessment) and motor (Movement Disorder Society Unified Parkinson’s Disease Rating Scale part 3) function at a baseline assessment, at an average of 1.3 years after diagnosis. We used logistic regression to determine predictors of PD with mild cognitive impairment and PD with dementia.
Results We studied 1893 patients with PD: 48 (2.5%) were heterozygous carriers for known Gaucher’s disease (GD) causing pathogenic mutations; 117 (6.2%) had non-synonymous variants, previously associated with PD, and 28 (1.5%) patients carried variants of unknown significance in the GBA gene. L444P was the most common pathogenic GBA mutation. Patients with pathogenic GBA mutations were on average 5 years younger at disease onset compared with non-carriers (P=0.02). PD patients with GD-causing mutations did not have an increased family risk of PD. Patients with GBA mutations were more likely to present with the postural instability gait difficulty phenotype compared with non-carriers (P=0.02). Patients carrying pathogenic mutations in GBA had more advanced Hoehn and Yahr stage after adjustment for age and disease duration compared with non-carriers (P=0.005). There were no differences in cognitive function between GBA mutation carriers and non-carriers at this early disease stage.
Conclusions Our study confirms the influence of GBA mutations on the age of onset, disease severity and motor phenotype in patients with PD. Cognition did not differ between GBA mutation carriers and non-carriers at baseline, implying that cognitive impairment/dementia, reported in other studies at a later disease stage, is not present in recently diagnosed cases. This offers an important window of opportunity for potential disease-modifying therapy that may protect against the development of dementia in GBA-PD.
Clinical trial registration NCT02881099; Results.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Initial reports of an association between some types of Gaucher’s disease (GD) and parkinsonism led to the investigation of the link between heterozygous mutations in the gene coding for the enzyme glucocerebrosidase (GBA) and Parkinson’s disease (PD).1 2 GBA mutations are common in PD and are the most important risk factor yet discovered for PD.3 A multicentre study of GBA mutation carriers, spanning three continents, established that the OR for any GBA mutation in patients with PD versus controls without PD was about 5.4.4 Variants in GBA can influence the phenotype of PD.5 Mutations that are not commonly recognised as causing GD have been found in patients diagnosed with PD6 and in particular the GBA E326K variant may predispose to PD.7 As compared with patients who do not carry a GBA mutation, those with GBA mutations have been reported to present earlier, are more likely to have affected relatives and are more likely to have atypical clinical manifestations.4 Furthermore, mild and severe heterozygous GBA mutations can differentially affect the risk and the age at onset of PD.8 GBA mutation carriers with PD are on a trajectory to cognitive decline9 despite clinicopathological studies showing no statistically significant difference in Braak stages between GBA mutation carriers and sporadic PD controls.10
Motor progression rates differ in GBA mutation carriers with PD. The HR for progression to Hoehn and Yahr stage 3 is significantly greater in GBA mutation carriers with PD compared with non-carriers.11 There is also evidence to suggest that carriers of polymorphisms in GBA which are not generally considered to increase PD risk are at significantly increased risk of progression to Hoehn and Yahr stage 3.11 More recent data from a prospective longitudinal study suggest that GBA-associated patients with PD not only have more rapid progression of disease but also have reduced survival.12
Cognitive impairment is a common problem in the later stages of PD,13 but even in early-stage disease there are mild deficits that can be identified on formal cognitive testing.14 Cognitive impairment in PD exists on a spectrum from mild cognitive impairment in PD (PD-MCI) to dementia (PD-D). Further, some types of PD-MCI may be a harbinger of dementia which occurs in up to 80% of patients over 15–20 years.13 and recent studies have identified several genetic factors that influence this including ApoE, MAPT and SNCA.14 15 GBA mutation status is reported as an independent risk factor for cognitive impairment in patients with PD.16 17 Further, GBA variants are reported to associate with a distinct pattern of cognitive deficits in PD characterised by greater impairment in working memory, executive function and visuospatial abilities.18 The individual risk of dementia in PD patients with GBA mutations is increased sixfold in carriers compared with non-carriers.17 The HRs for progression both to dementia and Hoehn and Yahr stage 3 are significantly greater in GBA mutation carriers compared with non-carriers, and in fact mutations in GBA have also been found in cases with dementia with Lewy bodies (DLB), thus pointing towards a link between GBA, parkinsonism and dementia.19
This collection of evidence from smaller studies linking GBA mutation status to several clinical phenotypic characteristics including disease progression and survival in PD has implications for genetic counselling, clinical follow-up and stratification in future clinical trials.
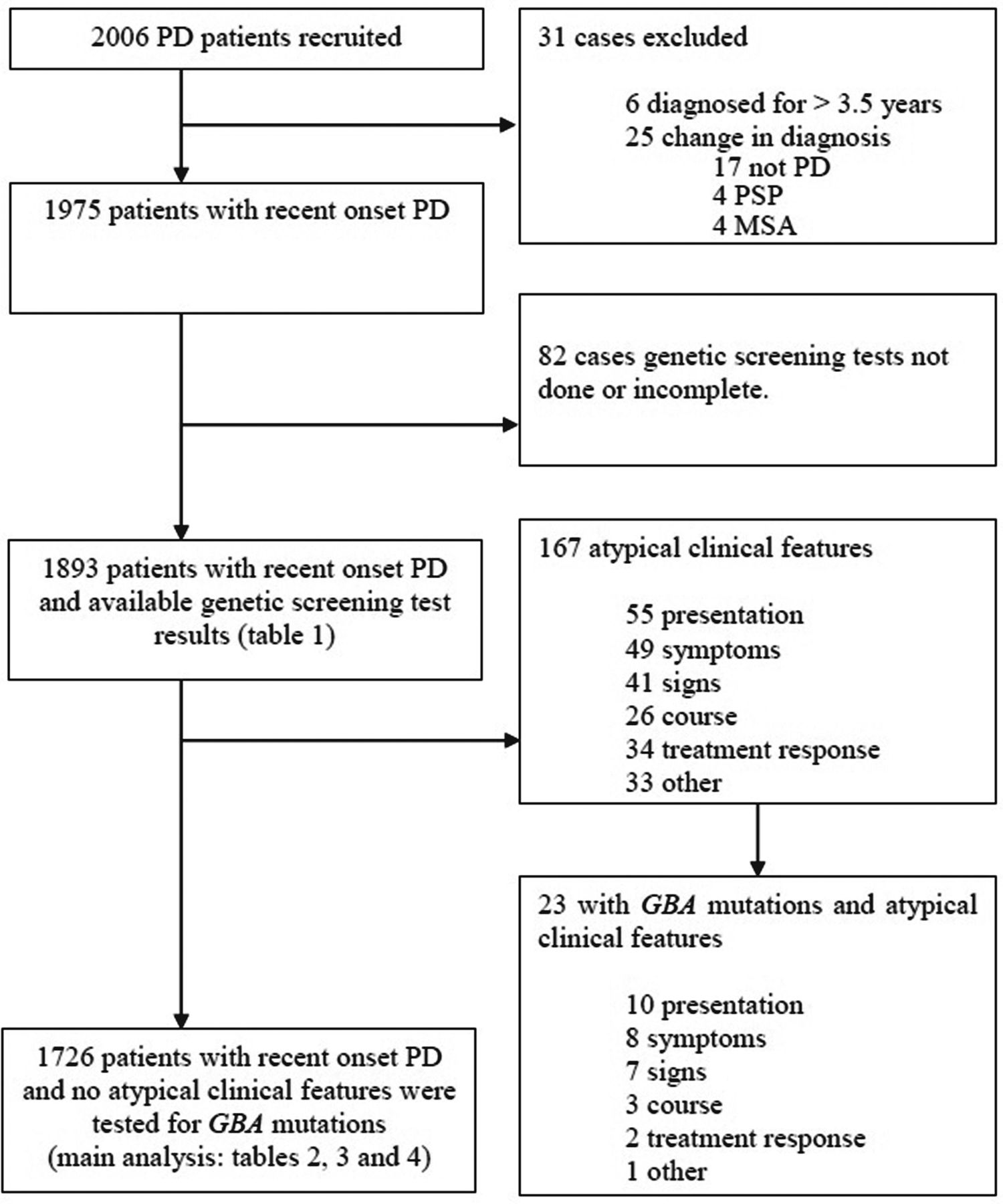
We assessed the influence of GBA mutations on the age of onset of PD, early motor phenotype, motor staging and cognitive function in a very large cohort of patients (n>2000, figure 1).
{kind=link}
Patient recruitment and assessment in this study. The main analysis of cognitive function using the Montreal Cognitive Assessment was performed in cases without features that might be atypical for Parkinson’s disease (PD). GBA, glucocerebrosidase PSA, Progressive supranuclear palsy, MSA, multiple system atrophy.
Methods
Tracking Parkinson’s is a large prospective, observational, multicentre project in the UK. The study set-up and design have been previously reported.20 Patients were recruited with a clinical diagnosis of PD, fulfilling Queen Square Brain Bank criteria21 and supported by structural and/or functional neuroimaging. Both drug-naive and treated patients aged 18–90 years were eligible. Recent onset cases were diagnosed with PD in the preceding 3.5 years, and recruitment was completed between February 2012 and May 2014. Patients with a clinical diagnosis of dementia at their first assessment were excluded. Enrolled patients whose diagnosis was later changed, on clinical or imaging grounds, were excluded from analysis. Additionally, patients with atypical features, which might indicate an alternative diagnosis, were excluded from the data analysis (figure 1).
Motor subtypes were determined from the Movement Disorder Society Unified Parkinson’s Disease Rating Scale part 3 (MDS-UPDRS 3) scores using a predetermined formula.22 The Montreal Cognitive Assessment (MoCA) test scores were adjusted for years of education and predetermined diagnostic cut-offs were used, to categorise cases into normal (>23), MCI 22–23 and dementia (<22), matching previous similar studies.20 23 Depression and anxiety were identified from scores >6 in the Leeds Hospital Anxiety and Depression Scale.24 Rapid eye movement (REM) sleep behaviour disorder (RBD) was defined as a score >4 on the RBD screening questionnaire.20 Olfaction testing was performed using either the 40-item University of Pennsylvania Smell Identification Test or Sniffin’ Sticks 16-item version, and hyposmia was defined as previously reported.25
Molecular genetic analysis of GBA mutations
The coding exons of GBA were screened using a nested PCR protocol followed by DNA sequencing described by Neumann et al.10 In accordance with the established convention for GBA alleles, all genetic variants were named according to the processed protein excluding the 39 residue signal peptide. Genetic variants were classified according to the criteria suggested by Winder-Rhodes et al11; GBA mutations that had been previously identified as being pathogenic in GD and associated with PD in the heterozygous state (group 1: e.g. L444P and G370S), non-synonymous genetic variants that have been linked to GD when occurring with other GBA mutations and have been associated with PD (group 2: eg, E326K and T369M) and genetic variants of unknown significance (group 3). Cases were then classified into one of these three groups depending on their GBA mutation status. Individuals with no sequence variants were classed as ‘non-carriers’ and combined with individuals from group 3. Further, analyses were performed comparing groups 1 and 2 with non-carriers of GBA mutations (group 3). Subgroup analyses were subsequently performed to compare cognition in p.L444P carriers to non-carriers and p.L444P mutation carriers to p.N370S carriers. Additional analyses were also performed to compare cognition in those with atypical features who carried GBA mutations and non-carriers.
Statistical analyses
Demographic and phenotypic characteristics were analysed using multivariable regression. Analyses of continuous variables (where UPDRS 3 and Parkinson’s disease questionnaire 8 items (PDQ8) scores were assumed continuous) were carried out using linear regression and binary variables using logistic regression. For MoCA scores, standard linear regression was used; for individual MoCA domains, ordered logistic regression was used. Where a category had small numbers (<3% of total), it was merged with the category above to ensure stability. Comparisons of Hoehn and Yahr stages were also carried out with ordered logistic regression. Comparisons of motor subtypes were performed using multinomial logistic regression with tremor dominant group as the comparator.
All P values were two-tailed; P values were calculated after adjustment for potential confounders where the confounders are listed in each table. The linearity of continuous confounders (such as age and disease duration) was tested using fractional polynomials in univariate models and then transformed if they showed evidence of non-linearity. We tested the proportional odds assumption in all ordered logistic regression models (except supplementary subgroup analyses with smaller numbers). All of these passed except within three models, and relaxing the model to a non-proportional odds model made no qualitative difference to the results.
Finally, we performed additional imputed analyses due to some missing data in the years of education (n=107), in the full MoCA test questionnaire (n=140) and within individual MoCA domains (varying between n=22–67 missing data elements). This involved using the chained equation approach to multiple imputation, creating 10 imputed datasets, then deriving estimates and P values with Rubin’s rules.
Statistical analyses were performed using STATA V. 13 (StataCorp).
Results
A total of 5 different pathogenic mutations, 28 variants of unknown significance (including two exon deletions) and 2 PD-associated non-GD variants (E326K, T369M) were found among 1893 patients with PD. Of these 1893 cases, 48 (2.5%) carried one or more GD pathogenic mutations; all of these cases were simple heterozygotes. In total, 117 carried PD-associated variants: 86 (4.5%) carried E326K mutation(s) (one case was homozygous for this mutation; the rest were heterozygous), 35 (1.8%) carried the T369M mutation (table 1) and 1728 were non-carriers of GBA mutations. Four carried more than one PD-associated variant. There were 167 cases with atypical features: 144 (86.2%) were non-carriers but 23 (13.8%) tested positive for GBA mutations. Of these 23 cases, 4 (2.4%) showed GD pathogenic variants while 19 (11.4%) had PD-associated non-GD variants (E326K or T369M). Detailed comparisons of phenotypic characteristics (tables 2–4) were performed on those without atypical clinical features that could raise suspicion of an alternative diagnosis (n=1726) (figure 1).
GBA variants found in the 1893 recently diagnosed patients from the Tracking Parkinson’s study
Demographic and clinical features of the Parkinson’s disease (PD) cohort classified by GBA mutation carrier status
Cognitive impairment in the GBA-associated Parkinson’s disease cohort classified by subdomain based on Montreal Cognitive Assessment (MoCA)
Cognitive and behavioural impairment in the GBA-associated Parkinson’s disease cohort based on the Movement Disorder Society Unified Parkinson’s disease rating scale part 1 scores dichotomised at 1 or above
Demographic and clinical features
In 142 PD cases who had a variant detected in the GBA gene (all cases), mean age was 65.6 years (SD 10.1) years, mean disease duration was 1.3 (SD 0.9) years and 93 (65.5 %) of these were male. Other demographic features of these cases classified by genotype (excluding cases with variants of unknown significance and those with unusual presentation) compared with non-carriers are detailed in table 2.
Cases who carried GD-causing variants in the GBA gene were younger (mean age 62.9, SD 12.3 years) compared with non-carriers (mean age 67.6 years, SD 9.2 years) (P<0.001), had an earlier age at onset (mean 59.7 years, SD 12.8 years) compared with non-carriers (mean 64.4 years, SD 9.7 years) (P=0.002) and were at a more advanced Hoehn and Yahr stage compared with non-carriers (P=0.02) when adjusted for age and disease duration (table 2). This was also reflected in greater medication requirements at baseline in those with GD-causing variants (levodopa-equivalent daily dose (LEDD) 370 mg/day, SD 219 mg/day) compared with non-carriers (LEDD 292 mg/day, SD 206 mg/day) (P=0.03). The postural instability gait difficulty (PIGD) motor subtype was the most common phenotype in those with GD-causing variants compared with those with non-carriers (adjusted P=0.04) reflecting more axial involvement and less tremor. E326K and T369M carriers when analysed together with carriers of GD-causing variants also showed PIGD as the most common motor subtype compared with non-carriers (adjusted P=0.03). The prevalence of anxiety (adjusted P=0.44), depression (adjusted P=0.28) and cognitive impairment (adjusted P=0.84) was not different between those with GD-causing variants (and those carrying the E326K, T369M variants) and non-carriers.
There was no statistically significant difference in the prevalence of RBD symptoms between those with mutations in the GBA gene and non-carriers (table 2). However, olfactory loss was more prevalent in those carrying GD causing mutations than non-carriers (P=0.047).
Cognitive profiles based on MoCA testing
There was no difference in cognition as measured by average MoCA test score or proportion of patients with dementia between those with and without pathogenic GBA mutations (table 3). There was no difference in cognition between possibly atypical PD cases carrying GBA mutations and PD non-carriers. (online supplementary table 1). Compared with non-carriers, those with GD-causing variants had lower scores on the naming domain of the MoCA test when adjusted for age, gender, disease duration and years of education (adjusted P=0.01), which did not survive correction for multiple testing. There were no significant group differences between those with GD-causing variants, E326K and T369M carriers and non-carriers in the scores of other domains of cognitive function such as attention (adjusted P=0.18), orientation (adjusted P=0.28), language (adjusted P=0.37) and delayed recall (adjusted P=0.67). However, E326K and T369M carriers had significant lower scores on the abstraction domain compared with those carrying GD-causing variants (adjusted P=0.02).
Supplementary file 1
Repeating the analyses for motor features, cognitive impairment and behavioural domains did not change the results.
Cases with p.L444P mutations did not show any difference in total MoCA test scores or any of the subdomains except naming (with a very small absolute difference) with non-carriers (online supplementary table 1). Cases with p.L444P mutations did not show any difference in any of the subdomains or total MoCA test scores compared with p.N370S mutation carriers (online supplementary table 2).
The data available from the answers to the six questions from part 1 of the MDS-UPDRS scale provide a measure of the impact of cognitive dysfunction and complex behaviours on the ability to perform activities of daily living as shown in table 4.
Finally, the imputed analyses showed no qualitative differences between the imputed and complete case results (online supplementary tables 1-7).
Discussion
To our knowledge, this is the largest single cohort study examining the influence of GBA mutations in newly diagnosed PD in terms of age at onset, motor phenotype and cognitive functions. We found that GBA mutations affected age at onset of PD and the motor phenotype, with no apparent effect on any aspect of cognition at the early stages of the disease process (average disease duration 1.5 years). We excluded patients with dementia at baseline as this is necessary to define PD as per Queen Square Brain Bank criteria.21 This means that patients with GBA-associated DLB are not included in this study.
2.5% of our cohort carried one or more pathogenic mutations in the GBA gene. This is lower than the proportion of cases found in a meta-analysis of European studies (5.5%)26 and a North American study (4.4%)18 but closer to the rate of GBA mutations found in a meta-analysis of data from studies in the Chinese population (3.6%).27 Higher proportions in other studies probably reflect data from enriched populations due to a referral bias to specialist centres for genetic testing. Our study reflects data collected from the National Health Service hospitals that normally provide routine healthcare in the UK, reflecting a diverse unbiased population pool in the community. L444P was the most common pathogenic GBA mutation (1.6%) in patients with PD in our study. This is in keeping with the results of a previous study from China that showed L444P as the most common mutation detected in GBA in cases with PD (n=402).27 This is not surprising given that L444P is a common pan-ethnic mutation while N370S is the most common mutation in Jewish populations.4
Some have suggested that GBA mutation carriers are more likely to have a family history of PD28 but we did not find any significant differences in the proportion of people with or without GBA mutations and a family history of PD among first-degree or second-degree relatives. We did not directly ask our participants whether they had Jewish ancestry but <1% reported that their religion was Judaism even though we included study centres in UK populations with large Jewish communities: North London, Manchester and Glasgow.
We found E326K in 4.5% of our patients with PD, which is close to the 4.7% figure reported in a cohort of similar size (n=1369) from the USA.18 The background control frequency of E326K in the UK has been reported to be approximately 2.5%. In our series, E326K was not associated with early-onset PD. One case in our series was homozygous for the E326K variant and had an age of onset 64.6 years and had similar motor features to E326K and T369M heterozygote carriers (group 2). This homozygous E326K case did not have GD, confirming that homozygosity for this allele does not cause GD, as described by others.7 One recent study reported the frequency of the E326K polymorphism in their cohort of patients with PD was no different from their controls , suggesting that E326K was not pathogenic for PD.29 However, results from a meta-analysis of data from five recent PD genome-wide association studies indicated that E326K is a susceptibility allele for PD.30
We found that our cases with GBA mutations were younger at diagnosis compared with non-carriers by about 5 years. Our data are consistent with a study from North America that reported similar findings.31 Another study from Germany similarly showed the age of onset in GBA mutation carriers was on average 6 years earlier compared with non-carriers,12 and a previous meta-analysis of several studies showed GBA mutation carriers had an earlier age of onset of around 4.6 years compared with PD patients without GBA mutations.32
We confirm previous reports suggesting that GBA variants can influence the motor phenotype of patients with PD.32 Those with GBA mutations in our study were at a more advanced Hoehn and Yahr stage, even when adjusted for disease duration, compared with non-carriers, which is consistent with previous reports.11 However, the UPDRS motor scores in our study were not significantly different between the groups, although all our patients were assessed ‘on’ medication. Those with GBA mutations (including E326K and T369M) were more likely to have the PIGD phenotype compared with non-carriers and had higher dopaminergic therapy requirements compared with non-carriers, possibly explaining the lack of difference in UPDRS motor scores. This was not reported by Winder-Rhodes et al in their mutation carriers (n=9),11 possibly because differences can be difficult to detect with small numbers of patients.
Earlier reports found that GBA variants are associated with greater impairment in working memory, executive function and visuospatial abilities18 and GBA mutations confer a greater risk of dementia during the course of PD.17
The development of therapeutic agents that exploit the key pathological pathways in GBA-associated PD include the small molecular chaperone Ambroxol,33 which crosses the blood–brain barrier when administered orally, have been shown to increase GBA enzyme activity in primate models.34 Therefore, if such therapeutic strategies were to become available for humans, we would need to identify a window of opportunity before the onset of dementia in GBA-associated PD where such interventions would have the maximum impact. Those earlier studies were reporting differences in cognitive function at an average disease duration of 12–15 years,16 17 whereas we are presenting baseline data.
Our findings are consistent with other previous studies that describe patients at this relatively early stage of the disease course (<3.5 years from disease onset) in which they did not find differences in cognitive function between patients carrying GBA mutations and non-carriers.9 11 35 Importantly, in those studies where cognitive scores were assessed longitudinally, patients carrying GBA mutations only begin to show divergence in cognitive performance around 36 months after motor onset (or diagnosis).9 11
The cognitive measures used in our study are restricted to the MoCA test. It is a widely validated test with sensitivity to detect changes in several cognitive domains for screening all levels of cognition in PD.23 Sensitivity and specificity for PD dementia using MoCA are 81%–82% and 75%–95%, respectively. Sensitivity and specificity for MCI in PD are 83%–90% and 53%–75%, respectively.23 We did not detect any differences at early-stage disease between those with pathogenic mutations in GBA and non-carriers in the attention, orientation, language, delayed recall and visuospatial domains using the MoCA test. While the MoCA has good sensitivity for detecting dementia and is a test of global cognition, subtle differences in individual cognitive domains may not be detected using the MoCA test even where they exist. Future studies could specifically address this issue with more sensitive neuropsychological measures in patients with specific mutations in order to detect more subtle cognitive deficits that may be found at an earlier stage.
Previous reports from genotype–phenotype correlative studies have suggested that those with p.L444P mutations (which are thought to cause a more severe GD phenotype) have a greater degree of cognitive impairment compared with patients with p.N370S mutations (which are thought to cause a less severe GD phenotype).8 We did not detect any differences in the cognitive performance on the MoCA test at this early stage in the disease in patients with these 2 types of mutations.
Besides cognitive impairment, previous reports suggest more severe anxiety and depression in GBA-associated PD compared with those with sporadic PD.35 36 The prevalence of anxiety in our study was not significantly different in pathogenic GD mutation carriers compared with non-carriers, and the same was true for depression Furthermore, we did not find any significant differences between the prevalence of neurobehavioural and psychiatric symptoms such as psychosis between the three groups of patients in our study. This may relate to the relatively early stage of disease in our cohort with a relatively shorter disease duration as psychosis in PD typically occurs in the later stages of the disease and after several years of treatment37 38 and differences may emerge with longer follow-up duration.
Strengths of our study include comprehensive screening of the GBA gene as opposed to selective mutation detection; the size of the study and the representative population base. Weaknesses of our paper include the cross-sectional family history data and the use of a brief cognitive screening tool.
Conclusion
We show that patients with PD and GBA mutations are different from GBA mutation non-carriers. GBA mutation carriers had an earlier age of onset, more commonly had the PIGD phenotype and had more severe motor impairment. In line with previous studies, global cognition scores did not differ between GBA mutation carriers and non-carriers at early stages in the disease process. Follow-up data emerging from this study will be used to reassess cognitive function in GBA mutation carriers at a later stageas well as the development of any neuropsychiatric and behavioural problems.
Acknowledgments
None
References
Footnotes
Contributors NM: data collection, manuscript writing and editing. RSW: manuscript writing and editing. MAL: data analysis, manuscript writing and editing. NB, RAB and TF: data collection and manuscript editing. JH and NWW: study design. CB and NWW: genetic analysis and manuscript editing. DJB: study design, data collection and manuscript editing. YB-S: study design, data analysis plan and manuscript editing. HRM, KAG and DGG: study design, data collection, analysis, manuscript writing and editing.
Funding The research was funded by Parkinson’s UK and supported by the National Institute for Health Research (NIHR) DeNDRoN network, the NIHR Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University, and the NIHR funded Biomedical Research Centre in Cambridge.
Disclaimer The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests YB-S has received grant funding from the MRC, NIHR, Parkinson’s UK, NIH and ESRC. RSW is supported by a UCL Excellence Fellowship and has received funding from the Academy of Medical Sciences and the NIHR, the MRC and the Wellcome Trust. NB has received payment for advisory board attendance from UCB, Teva Lundbeck, Britannia, GSK, Boehringer and honoraria from UCB Pharma, GE Healthcare, Lily Pharma, Medtronic. He has received research grant support from GE Healthcare, Wellcome Trust, Medical Research Council, Parkinson’s UK and National Institute for Health Research. RAB has received grants from Parkinson’s UK, NIHR, Cure Parkinson’s Trust, Evelyn Trust, Rosetrees Trust, MRC and EU along with payment for advisory board attendance from Oxford Biomedica LCT and FCDI and honoraria from Wiley and Springer. DJB has received grants from NIHR, Wellcome Trust, GlaxoSmithKline Ltd, Parkinson’s UK and Michael J Fox Foundation. JH has received honoraria from Eisai and grant support from MRC/Wellcome, Parkinson’s UK and the Michael J Fox Foundation. DGG has received grants from Michael’s Movers, The Neurosciences Foundation, and Parkinson’s UK, and honoraria from UCB Pharma and GE Healthcare, and consultancy fees from Acorda Therapeutics. HRM has received grants from Medical Research Council UK, Wellcome Trust, Parkinson’s UK, Ipsen Fund, Motor Neurone Disease Association, Welsh Assembly Government, PSP Association, CBD Solutions and Drake Foundation, and payment for advisory board attendance and lectures from Teva, AbbVie, Boehringer Ingelheim, and GSK. NWW is supported by the MRC and NIHR UCLH Biomedical research centre. TF has received grants from Michael J Fox Foundation, Cure Parkinson’s Trust, Brain Research trust, John Black Charitable Foundation, Rosetrees trust and honoraria for speaking at meetings from Bial, Profile Pharma and Medtronic.
Patient consent Obtained.
Ethics approval West of Scotland Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.