Article Text

Abstract
Objectives Cognitive and behavioural changes within the spectrum of frontotemporal dementia (FTD) are observed frequently in patients with amyotrophic lateral sclerosis (ALS). Whether these changes also occur in other forms of motor neuron disease (MND) is not well studied. We therefore systemically screened a large cohort of patients with primary lateral sclerosis (PLS) and progressive muscular atrophy (PMA) for cognitive and behavioural changes, and subsequently compared our findings with a cohort of patients with ALS.
Methods Using a set of screening instruments (Edinburgh Cognitive and Behavioural ALS Screen, ALS and Frontotemporal Dementia Questionnaire, Frontal Assessment Battery, and Hospital Anxiety and Depression Scale), the presence of cognitive and behavioural changes as well as anxiety and depression in 277 patients with ALS, 75 patients with PLS and 143 patients with PMA was evaluated retrospectively.
Results We found a high frequency of cognitive and behavioural abnormalities with similar profiles in all three groups. Subjects with behavioural variant FTD were identified in all groups.
Conclusions The percentage of patients with PLS and PMA with cognitive dysfunction was similar to patients with ALS, emphasising the importance for cognitive screening as part of routine clinical care in all three patient groups. With a similar cognitive profile, in line with genetic and clinical overlap between the MNDs, the view of PLS as an MND exclusively affecting upper motor neurons and PMA exclusively affecting lower motor neurons cannot be held. Therefore, our findings are in contrast to the recently revised El Escorial criteria of 2015, where PLS and PMA are described as restricted phenotypes. Our study favours a view of PLS and PMA as multidomain diseases similar to ALS.
Statistics from Altmetric.com
Introduction
Motor neuron disease (MND) is a group of several neurodegenerative disorders thought to selectively affect the motor neurons. However, over the past decade it has become increasingly clear that in the most common form of MND, amyotrophic lateral sclerosis (ALS), neurodegeneration is not limited to motor neurons and that other parts of the brain are affected as well (mainly the frontal and temporal lobes). Up to 50% of patients with ALS have some degree of cognitive and/or behavioural impairment, and up to 15% have concomitant frontotemporal dementia (FTD).1 Moreover, ALS and FTD are considered the phenotypic extremes of the same disease, the FTD-MND continuum.2
Whether pathology extends outside of the motor system in other forms of MND, such as primary lateral sclerosis (PLS) and progressive spinal muscular atrophy (PMA), has not been studied extensively. There are a few small studies and case reports/series suggesting that cognitive and behavioural changes as well as FTD may be seen in PLS.3 The literature on PMA is very limited and conflicting, although cognitive involvement has been reported.4 5 The latest diagnostic criteria (revision of the El Escorial criteria of 2015) however consider PMA and PLS to be restricted phenotypes,6 meaning that in PMA the loss of motor neurons occurs exclusively in the spinal cord and that in PLS neurodegeneration is limited to the motor cortex.
In this study we investigated if PMA and PLS are indeed restricted phenotypes or whether there is more widespread involvement of the nervous system by systematically screening large cohorts of patients with PLS and PMA for cognitive and behavioural changes, and subsequently compared our findings with a cohort of patients with ALS.
Methods
Subjects
A total of 75 patients with PLS, 143 patients with PMA and 277 patients with ALS were included in this study. These patients were recruited from the outpatient clinic of the neurology department at the University Medical Center Utrecht, which is the national referral centre of MND in the Netherlands. Retrospectively, we included both incident and prevalent cases that were seen between 2014 and 2017. Patients with PLS were diagnosed according to the Gordon criteria,7 and patients with PMA fulfilled previously described criteria.8 Patients with ALS were diagnosed with possible, probable, probable laboratory-supported or definite ALS according to the revised El Escorial criteria.6 Cognitive and behavioural screening was performed either as part of the regular diagnostic work-up at our outpatient clinic or in ongoing research projects.9 Patients had to fulfil the following inclusion criteria: (1) Dutch as the first language; and (2) the absence of pre-existing conditions that could influence test performance: dyslexia, learning disabilities, substance abuse, psychiatric disorders, other neuromuscular diseases, cerebrovascular disease, epilepsy, neurodegenerative diseases, traumatic brain injury and/or the use of psychoactive medication.
Cognitive and behavioural assessment
The following validated screening instruments were used: Edinburgh Cognitive and Behavioural ALS Screen (ECAS),10 the ALS and Frontotemporal Dementia Questionnaire (ALS-FTD-Q),11 the Frontal Assessment Battery (FAB),12 and the Hospital Anxiety and Depression Scale (HADS).13
Cognitive functions were assessed using the Dutch version of the ECAS, which is a brief multidomain screening tool that assesses functions typically affected in ALS (ALS-specific part: fluency, executive functions, language and social cognition) as well as functions not commonly affected in ALS (ALS non-specific part: memory and visuospatial functions, which are more frequently affected in other disorders of older adults). Education and age adjusted cut-off values were applied. The limit of the normal range was defined as the fifth percentile (LA Bakker et al, submitted).
The FAB is a screening battery which is sensitive to frontal lobe dysfunction and consists of six subtests assessing conceptualisation, mental flexibility, motor programming, sensitivity to interference, inhibitory control and environmental autonomy. Scores below 12 are considered indicative of frontal dysfunction.14
Two caregiver interviews (the ECAS behavioural interview and the ALS-FTD-Q) were used to provide an assessment of behavioural changes and psychotic symptoms associated with FTD with MND. The ECAS behavioural interview is a semistructured interview based on the five key behavioural domains affected in behavioural variant FTD (bvFTD) and reflects the most recent diagnostic criteria,15 and also includes questions on psychotic symptoms associated with ALS-FTD.16
The ALS-FTD-Q is a paper questionnaire that contains 25 items assessing the most frequently observed behavioural symptoms in patients with ALS-FTD on a 4-point rating scale with a maximum score of 100. Scores on the ALS-FTD-Q ≤21 are considered as normal, scores ≥22 indicate mild behavioural changes and scores ≥29 indicate severe behavioural changes.11 Both the ECAS behavioural interview and the ALS-FTD-Q were administered to an informant/caregiver separately from the patient.
Finally, the HADS was used to exclude subjects with depression or anxiety disorders, as these conditions could influence both neuropsychological and behavioural test results. We excluded 101 patients with ALS, 4 patients with PLS and 9 patients with PMA based on a HADS score of ≥11 on the anxiety and/or depression subscale, indicating the likely presence of an anxiety disorder and/or depression.13
Classification of neuropsychological and behavioural profiles
We classified patients with cognitive and/or behavioural changes based on the revised diagnostic criteria for ALS-frontotemporal spectrum disorder (revised Strong criteria).17 In these criteria patients are considered to have the following:
Behavioural impairment (bi) if there was apathy with or without other behavioural changes or at least two non-overlapping supportive diagnostic features from the Rascovsky criteria.15
Cognitive impairment (ci) if there was executive dysfunction (including social cognition) or language dysfunction or a combination of the two. Executive impairment is defined as impaired verbal fluency (letter) or impairment on two other non-overlapping measures of executive functions (which may include social cognition). Language impairment is defined as impairment on two non-overlapping tests and in which language impairment is not solely explained by verbal fluency deficits.
Cognitive and behavioural impairment (cbi) when patients met the criteria for both ci and bi.
Comorbid FTD when there was evidence of progressive deterioration of behaviour and/or cognition by observation or history; AND (1) at least three of the Rascovsky criteria OR (2) at least two of the Rascovsky criteria in combination with loss of insight and/or psychotic symptoms OR (3) language impairment meeting the criteria for semantic variant primary progressive aphasia (PPA) or non-fluent variant PPA, which may coexist with other behavioural and/or cognitive symptoms as outlined above.
Amnestic mild cognitive impairment (aMCI) when patients scored below the fifth percentile on memory, but which did not interfere with activities of daily living and normal scores on all other domains. aMCI was not included in the revised Strong criteria, but is frequently seen in the ageing general population and viewed as an intermediate state between normal cognition and Alzheimer’s disease (AD).18
Comorbid dementia when patients fulfilled the diagnostic criteria for a dementia other than FTD: AD, vascular dementia19 or mixed dementia (eg, AD-vascular dementia).
Statistical analysis
Statistical analysis was carried out using SPSS V.22 statistical software. Comparisons between diagnostic groups on baseline demographics, clinical features, test scores, frequency of cognitive and behavioural changes as well as cognitive profiles were made using analysis of variance, independent t-test, χ2 test, Fisher’s exact test, Mann-Whitney U test and Kruskal-Wallis test as appropriate.
Results
Baseline characteristics
Comparison of demographics and clinical characteristics (table 1) showed that there were no differences in age at evaluation between the three groups. The PMA group consisted of significantly more men than the other two groups (p<0.05). Disease duration was significantly longer in the PLS group compared with both the ALS and the PMA groups (p<0.01). This is to be expected as the diagnostic criteria for PLS require patients to have upper motor neuron (UMN) signs for at least 4 years in the absence of lower motor neuron signs,20 whereas the average survival for patients with ALS is 3–4 years from symptom onset (and slightly longer for PMA).8 Moreover, ECAS data from patients with ALS came primarily from their initial visit to the outpatient clinic. Perhaps correlated to the longer disease duration, the Revised ALS Functional Rating Scale scores were significantly lower in the PLS group, indicating more advanced disease stage. Bulbar onset was more common in ALS than in PLS (29% vs 13%) and very rare in PMA (1 out 143 cases). Overall, the clinical characteristics appear to be in line with the literature, and we therefore consider these cohorts to be representative. Moreover, for PLS and PMA these are among the largest cohorts to be reported to date. Interestingly, the percentage of patients with PLS with a high level of education (International Standard Classification of Education categories 5 and 6) was significantly higher than in the ALS and PMA groups (33% vs 19% and 20%, respectively). Considering age and education adjusted cut-off values were applied, these differences in level of education did not influence the outcome of the study.
Demographic and clinical characteristics
Cognitive screening
In all three patient groups we found a relatively high percentage of abnormal neuropsychological performance on the ECAS, varying between 13% and 24%. The frequency of ALS-specific abnormal scores did not differ between the ALS, PLS and PMA groups (figure 1). Abnormal ECAS total and non-specific scores were more frequent in patients with ALS compared with PMA, which seems to be driven by memory impairment. Fluency, executive function, language and memory were most frequently affected (figure 2, online supplementary figure 1), which is a finding that mirrors the cognitive profile of ALS.17 Abnormal visuospatial function was uncommon in all three groups. The results of the FAB were very similar in ALS and PLS, with abnormal scores in 10% and 12% of subjects, respectively, but found in only 1 patient with PMA (2%).
Supplemental material

Percentage of subjects with an abnormal score on the ECAS, FAB and ALS-FTD-Q. *P<0.05. Availability of data: FAB: ALS 100%, PLS 45%, PMA 31%; ALS-FTD-Q: ALS 100%, PLS 60%, PMA 62%. ALS, amyotrophic lateral sclerosis; ALS-FTD-Q, ALS and Frontotemporal Dementia Questionnaire; ECAS, Edinburgh Cognitive and Behavioural ALS Screen; FAB, Frontal Assessment Battery; PLS, primary lateral sclerosis; PMA, progressive muscular atrophy.
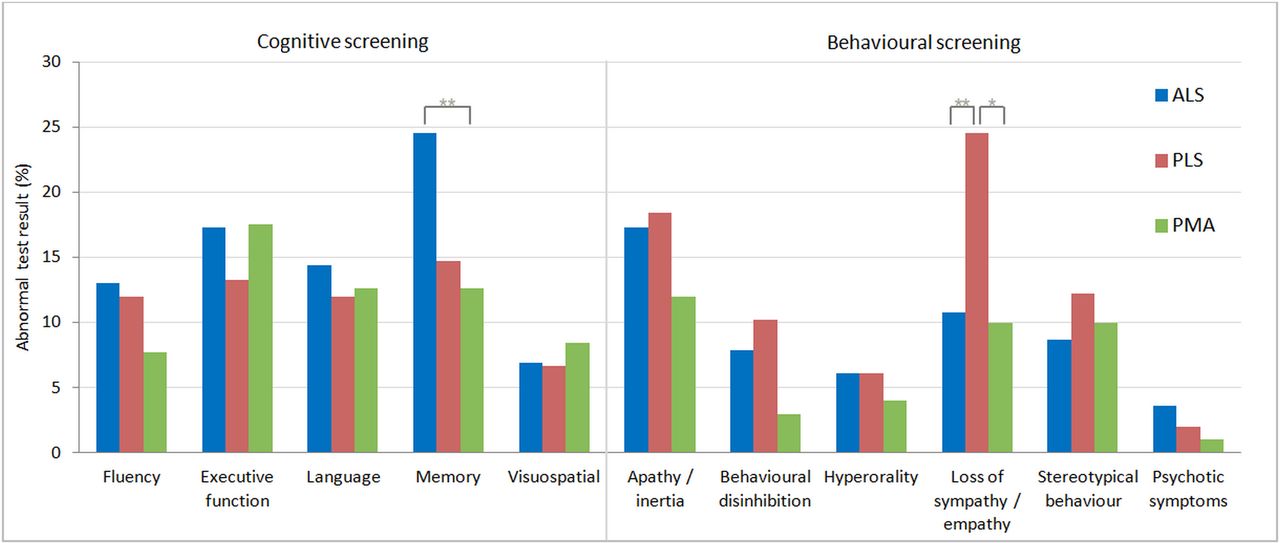
Abnormal performance per ECAS cognitive and behavioural domain. *P<0.05, **p<0.01. Available ECAS behavioural interview: ALS 100%, PLS 65%, PMA 70%. ALS, amyotrophic lateral sclerosis; ECAS, Edinburgh Cognitive and Behavioural ALS Screen; PLS, primary lateral sclerosis; PMA, progressive muscular atrophy.
Behavioural screening
The results from the ECAS behaviour screen and ALS-FTD-Q identified behavioural changes in all three patient groups. The three groups have a similar behavioural profile with a trend towards a lower percentage and less severe behavioural symptoms in PMA. Of the five assessed behavioural domains, loss of sympathy was more frequently reported in PLS (25%) than in ALS and PMA. Psychotic symptoms were rare in all groups (figures 1 and 2).
Classification of neuropsychological and behavioural profiles
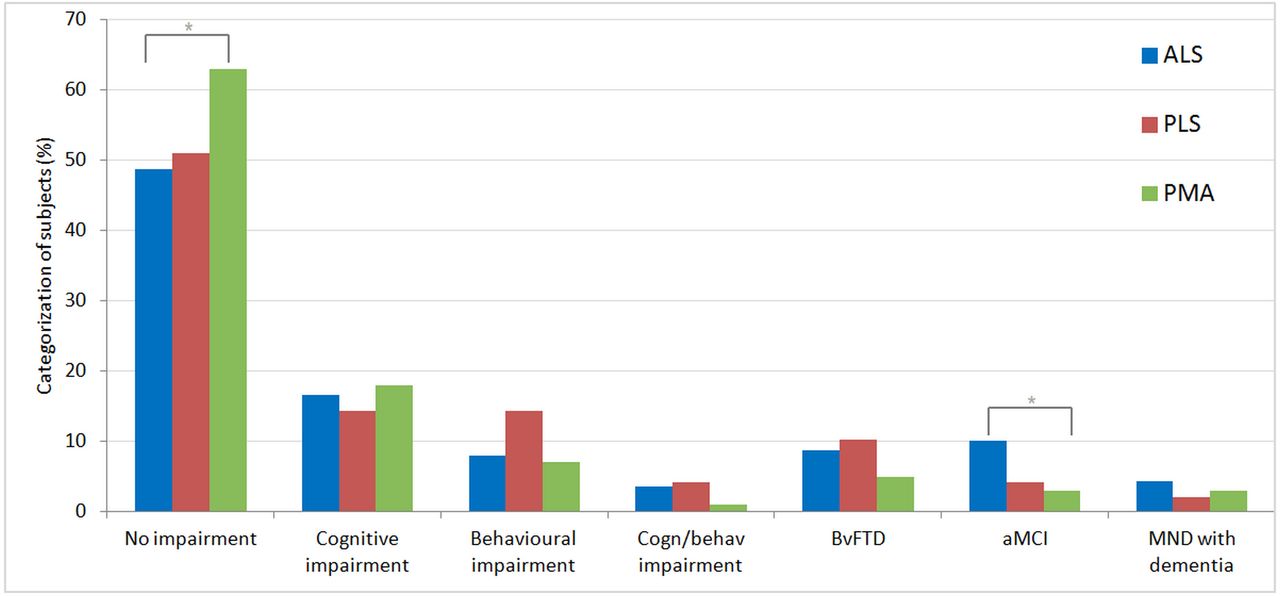
Classification of subjects according to the revised Strong criteria17 is shown in figure 3. Similar to the findings based on individual tests, application of the revised Strong criteria shows the highest percentage of individuals without abnormalities in the PMA group. Cognitive impairment was seen at equal frequencies across all groups, whereas behavioural changes (bi or bvFTD) appear to be more common in ALS and PLS. Cognitive impairment without behavioural features (ci) is found most often in the PMA group. Interestingly, a considerable number of subjects with abnormal behaviour (bi, bvFTD) scored normally on all cognitive domains, and vice versa (online supplementary table 1). There was a trend towards a higher frequency of bvFTD in C9orf72-positive ALS cases versus ALS cases without the expansion (16.7% vs 8.7%, p=0.16). Psychotic features were uncommon in all patient groups. We observed psychotic features in 1 out of 18 C9orf72-positive patients with ALS compared with 5 out of 178 C9orf72-negative patients (5.6% vs 2.8%, p=0.44).
Supplemental material
{kind=link}
{kind=link}
{kind=link}
Categorisation in cognitive and behavioural profiles. *P<0.05. Classification is according to the revised Strong criteria,17 with the addition of the category aMCI.18 ALS, amyotrophic lateral sclerosis; aMCI, amnestic type mild cognitive impairment; behav, behavioural; cogn, cognitive; bvFTD, behavioural variant frontotemporal dementia; MND, motor neuron disease; PLS, primary lateral sclerosis; PMA, progressive muscular atrophy.
Discussion
ALS is increasingly viewed as a neurodegenerative syndrome with diverse clinical manifestations (rate of disease progression, age of onset, presence of cognitive and behavioural changes, and so on) caused by multiple underlying pathophysiological processes resulting from mutations in over 20 genes, environmental exposures or both.21 The concept of a syndrome rather than a single disease is perhaps even more applicable to PMA. Over time its definition has changed following the identification of diseases such as multifocal motor neuropathy, Hirayama disease, Kennedy’s disease and distal spinal muscular atrophy. Despite these significant advances, PMA likely still represents a heterogeneous group of disorders, although with increasing similarity to ALS. In fact, whether PMA is a form of ALS is a long-standing debate that dates back to the days of Charcot (who proposed a sharp division between the two) and Dejerine (who advocated lumping).22 Although PLS is a relatively well-defined clinical syndrome characterised by the progressive loss of UMNs, differentiating PLS from (sporadic) hereditary spastic paraplegia (HSP) in patients with lower limb onset or from an UMN-predominant presentation of ALS may be challenging.23 24 Similarly, there is a debate on whether PLS should be viewed as a subtype of ALS.
The current consensus criteria consider PLS and PMA as restricted phenotypes of ALS, in which motor neuron degeneration is limited to either the motor cortex or spinal cord, respectively. They were kept as subcategories in the revised criteria as the prognosis differs from classical ALS. Patients with clinically ‘pure’ UMN or lower motor neuron (LMN) phenotypes have slower disease progression compared with ALS.6
Several small studies and case reports/series have however suggested that cognitive and behavioural changes may occur in PMA and PLS, which implies that motor neuron degeneration is in fact not anatomically restricted to the spinal cord or motor cortex.3–5 We therefore set out to systematically investigate cognitive and behavioural function in a large cohort of patients with PMA and PLS (and ALS for comparison). We show that cognitive and behavioural changes are indeed common in these disorders. When classifying according to the revised Strong criteria, we found that 37% of patients with PMA and 49% of patients with PLS have some degree of cognitive and/or behavioural abnormality.
The most commonly affected cognitive domains in PLS and PMA were executive function, language, memory and fluency. Although memory deficits were relatively common, this was rarely found to be affected first, in isolation or the most severely affected domain. A recent meta-analysis on the cognitive profile of FTD indeed also shows that verbal memory (not episodic memory) is perhaps more frequently affected in FTD than commonly thought.25 Therefore, we did not consider these patients to have unrelated, coincidental cognitive decline.
The cognitive and behavioural changes in PLS and PMA are in keeping with frontotemporal dysfunction. Moreover, the cognitive profile of patients with PMA and PLS was highly similar to that of the patients with ALS in this study, with only small differences (more common memory impairment in ALS and loss of sympathy in PLS). Our findings in all three groups are also in line with the results from the latest meta-analysis on the cognitive profile of ALS, which also describes deficits in fluency, executive function, language and memory.26
Diagnostic criteria commonly undergo revision as new insights emerge. In this study we show that cognitive and behavioural changes are common in PLS and PMA. Neurodegeneration in these diseases is not limited to a specific population of motor neurons, but is more widespread. We therefore feel that the concept of PLS and PMA as restricted phenotypes of ALS is unattainable.
The strikingly similar cognitive profiles to ALS, however, do support viewing PMA and PLS as (sub)types of ALS. Our findings add to a growing body of evidence in favour of lumping these disorders. Under current diagnostic criteria, patients with PMA phenotypes and a positive family history for ALS are considered to have ALS.6 It has also been shown that patients with apparently sporadic PMA carry mutations in the ALS genes such as SOD1, ANG, FUS/TLS, TARDBP, CHMP2B and C9orf72.27–29 Similarly, mutations in C9orf72, DCTN1 and TBK1 have been identified in patients with PLS.28 30 Additionally there are reports of patients with PLS with a positive family history for ALS.31
The number of histopathological studies in PMA and PLS is limited, but also suggests that neurodegeneration is not restricted. In PMA corticospinal tract degeneration was observed in roughly half of the patients that came to autopsy.32 Those in favour of viewing PMA as a form of ALS often argue that severe loss of lower motor neurons can mask the presence of UMN signs and therefore complicates making a diagnosis of ALS in these cases. Indeed, some patients with PMA do develop clear UMN signs over time and thus convert to ALS. Although detecting UMN signs in the presence of severe weakness and muscle wasting will remain challenging, screening for cognitive and behavioural changes is a simple and effective way of showing involvement of the central nervous system.
In patients with PLS mild neuronal loss in the brainstem nuclei as well as the presence of bodies and ubiquitin inclusions have also been reported,33 as well as involvement of the frontal and temporal lobes.3 Moreover, several studies have shown TAR DNA-binding protein 43 (TDP-43) pathology in patients with PLS and PMA, which is the pathological hallmark of ALS and FTD.34 35
Recognising PMA as PLS as a subtype of ALS is highly relevant from both a clinical and scientific perspective. In the clinic detecting cognitive and behavioural changes may aid in the diagnostic process. Differentiating PMA from other neuromuscular diseases with pure lower motor neuron signs may be challenging, in particular in atypical cases. However, these mimics solely affect the peripheral nervous system leaving cognition intact. Similarly, the identification of cognitive changes may help differentiate between pure sporadic HSP and PLS.
Cognitive/behavioural impairment should also be taken into account in the clinical management as ALS with signs of FTD is associated with non-compliance with treatment recommendations, affects medicolegal decision making, negatively influences survival and significantly increases caregiver burden.36 At present little is known about the timing at which cognitive and behavioural changes may occur in PMA and PLS. We previously however reported several PLS cases in which full-blown FTD developed within months, after years of exclusive motor deficits and without an increase in the pace of motor deterioration.3 We would therefore advocate performing cognitive and behavioural screening as part of the diagnostic process as well as at regular intervals during follow-up.
Viewing PMA and PLS as forms of ALS may potentially open doors to these patients from a treatment perspective. The rare nature of PMA and PLS makes therapy development for these disease specifically highly challenging. At present patients with PLS and PMA are also excluded from participating in ALS trials, given the ongoing debate on the nature of these diseases. Therefore, current treatment in PLS and PMA is primarily supportive and symptomatic. However, the increasing evidence of genetic, phenotypic and pathological overlap with ALS would seem to justify offering drugs that are effective in ALS to patients with PMA and PLS as well, in particular in the event that gene-specific therapy should become available.
There are different hypotheses with regard to the onset and progression of ALS. It has been suggested that neurodegeneration occurs via a dying-back mechanism, in which after the death of a neuron the connecting axon secondarily degenerates.37 The autopsy findings in PMA (severe loss LMNs with mild degeneration of the corticospinal tract)32 and the autopsy findings and our results in PLS (frequent frontotemporal involvement in the absence of LMN signs) would support dying-back.38 However, the identification of cognitive and behavioural changes in patients with PMA with no UMN involvement would argue for a multifocal process, rather than spread from a point of focal through neuroanatomically connected pathways (be it via dying-back, prion-like mechanisms or other).37 39 Considering the heterogeneity of the MNDs, it is also entirely possible that different mechanisms are at play in various subtypes. It seems however that further studying PMA and PLS could provide window into the mechanisms of spread and potentially cell-specific vulnerability.
Our study has several limitations. In a large proportion of patients with PLS and PMA (52% and 71%, respectively), the HADS results were missing. This might have resulted in the inclusion of subjects with significant depressive and/or anxiety symptoms, potentially influencing test results. However, we did not observe more cognitive impairment in subjects lacking HADS data compared with patients with HADS data. Lastly, although the screening instruments we used in this study assess language, it is important to note that these language tests may lack sufficient detail to detect and/or distinguish the language variants of FTD: semantic variant PPA or non-fluent variant PPA. Therefore it is possible that we underestimate the number of subjects with (language variant) FTD in our cohort.
Conclusions
Our results indicate that both cognitive and behavioural impairments, including FTD, are common in ALS and in PLS and PMA. We found a highly similar cognitive and behavioural profile in the three MNDs, with a trend of less frequent abnormal findings in PMA. Our results emphasise the importance of cognitive and behavioural screening as part of routine clinical care in all three MND patient groups. With a similar cognitive profile, in line with pathological, imaging, familial, genetic and clinical evidence for overlap between the MNDs, the view that PLS is an MND exclusively targeting the UMNs and PMA exclusively targeting the lower motor neurons cannot be held. Therefore, our findings are in contrast to the recently revised El Escorial criteria of 2015, where PLS and PMA are described as restricted phenotypes. We believe that PLS and PMA are multidomain diseases similar to ALS, or more likely subtypes of ALS.
References
Footnotes
Contributors MAvE and TCWN were responsible for the study concept. BSdV, LMMR, CDS, JHV and LHvdB contributed substantially to data acquisition and interpretation of the analysis. BSdV, LMMR, LAB, TCWN and MAvE analysed the data. BSdV and MAvE drafted the manuscript, which was reviewed, edited and approved by all the authors. MAvE is the guarantor of the overall content.
Funding MAvE receives funding from the Netherlands Organization for Health Research and Development (Veni scheme), The Thierry Latran Foundation, the ALS Foundation Netherlands and JPND.
Competing interests MAvE serves on the UK Motor Neurone Disease Association Biomedical Research Advisory Panel, has consulted for Biogen and has received travel grants from Shire (formerly Baxalta). LHvdB reports grants from Netherlands ALS Foundation, grants from Prinses Beatrix Spierfonds, grants from Netherlands Organization for Health Research and Development (Vici scheme), and grants from European Community’s Health Seventh Framework Programme (FP7/2007-2013) (grant agreement no 259867), during the conduct of the study; and personal fees from Baxter for scientific advisory board and travel grant, and personal fees from Scientific Advisory Board, Biogen Idec, outside the submitted work. The other authors declare that they have no conflict of interest.
Patient consent Not required.
Ethics approval The study was approved by the institutional review board of the UMCU.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary