Article Text

Abstract
The frontotemporal dementia (FTD) spectrum is a heterogeneous group of neurodegenerative syndromes with overlapping clinical, molecular and pathological features, all of which challenge the design of clinical trials in these conditions. To date, no pharmacological interventions have been proven effective in significantly modifying the course of these disorders. This study critically reviews the construct and methodology of previously published randomised controlled trials (RCTs) in FTD spectrum disorders in order to identify limitations and potential reasons for negative results. Moreover, recommendations based on the identified gaps are elaborated in order to guide future clinical trial design. A systematic literature review was carried out and presented in conformity with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses criteria. A total of 23 RCTs in cohorts with diagnoses of behavioural and language variants of FTD, corticobasal syndrome and progressive supranuclear palsy syndrome were identified out of the 943 citations retrieved and were included in the qualitative review. Most studies identified were early-phase clinical trials that were small in size, short in duration and frequently underpowered. Diagnoses of populations enrolled in clinical trials were based on clinical presentation and rarely included precision-medicine tools, such as genetic and molecular testing. Uniformity and standardisation of research outcomes in the FTD spectrum are essential. Several elements should be carefully considered and planned in future clinical trials. We anticipate that precision-medicine approaches will be crucial to adequately address heterogeneity in the FTD spectrum research.
- frontotemporal dementia
- randomised trials
- systematic reviews
- pharmacology
- neurogenetics
Statistics from Altmetric.com
Introduction
The frontotemporal dementia (FTD) spectrum encompasses a heterogeneous group of neurodegenerative syndromes presenting with a wide range of overlapping clinical features. FTD represents the second most common type of early-onset dementia, approaching the prevalence of Alzheimer’s disease (AD) in the 45–64 years age group.1 2 It comprises two main clinical phenotypes: behavioural variant FTD (bvFTD), where behavioural changes and executive dysfunction are prominent early manifestations, and primary progressive aphasia (PPA), where comprehension and/or production of language are impaired.3 PPA is further divided according to the specific language deficits into non-fluent variant PPA (nfvPPA) and semantic variant PPA (svPPA). Another subtype of PPA, logopenic variant PPA, is more frequently associated with underlying Alzheimer’s pathology at autopsy.4–6 Other FTD spectrum disorders include the clinical phenotypes of progressive supranuclear palsy syndrome and corticobasal syndrome (CBS). Motor manifestations, such as bradykinesia, rigidity and dystonia, and cortical deficits such as apraxia are common manifestations during the course of these disorders.7 8 Finally, amyotrophic lateral sclerosis (ALS) is also strongly linked to the FTD spectrum (ie, frontotemporal dementia with motor neuron disease (FTD-MND)) as it shares common pathological findings and genetic mutations.9 About 10%–15% of patients with ALS meet the diagnostic criteria for FTD at baseline.10 While each of these disorders has distinctive features and can be differentiated clinically from one another, there can be overlapping clinical features between the classic clinical phenotypes of FTD spectrum disorders, thus complicating the clinical diagnostic picture.
These neurodegenerative syndromes also share common underlying anatomical, molecular and pathological substrates. Neuropathological examination of individuals with FTD spectrum disorders reveals findings of frontotemporal lobar degeneration (FTLD), where atrophy is prominent in the frontal and/or temporal lobes. In most cases, neuronal loss and gliosis are thought to be secondary to neuronal and astrocytic inclusions of microtubule-associated protein tau, TAR DNA-binding protein 43 (TDP-43), and more rarely RNA-binding protein fused in sarcoma (FUS).11–13 Behavioural and language variants of FTD are associated with tau, TDP-43 or FUS proteinopathies,14 and progressive supranuclear palsy syndrome is most often associated with tau proteinopathy, specifically progressive supranuclear palsy (PSP) pathology.7 8 While CBS can also be secondary to tauopathy in the form of corticobasal degeneration (CBD) or PSP, other proteinopathies, such as beta-amyloidopathy/tauopathy (ie, AD), prionopathy, TDP-43 proteinopathy and alpha-synucleinopathy, can also produce the syndrome.8 15
Genetic mutations inherited in an autosomal dominant fashion can cause FTD spectrum disorders, with 10%–20% of all cases attributed to mutations in or near three different genes: C9orf72 (encoding protein C9orf72), GRN (encoding progranulin) and MAPT (encoding microtubule-associated protein tau).11 14 16 Although more rare, other disease-causing genetic mutations have also been identified in heritable FTD, such as in VCP, TARDBP, TIA1, TBK1 and CCNF genes for cases of FTD due to TDP proteinopathy, and in CHMP2B and FUS for cases of FTD due to tau-negative, TDP-negative, ubiquitin-positive pathology.17–19 For PSP and CBD not due to MAPT mutations (ie, sporadic disease), common variation in MAPT, specifically the MAPT H1 haplotype, is an important genetic risk factor for these disorders.20 Variants tagging the MAPT H1 haplotype were not surprisingly confirmed in a genome-wide association study (GWAS) of PSP, but several novel common variants in the STX6, EIF2AK3 and SLC25A38/Appoptosin genes were found to increase the risk for this disease.21 22 With respect to sporadic TDP-43 proteinopathies and those caused by GRN mutations, common variants within the TMEM106B gene that increase its expression were found to increase the risk and were associated with shorter disease duration in FTD cases; the major allele (T) increased the risk and was associated with a shorter disease duration, while the minor allele (G) had protective effects.23 In GRN carriers, the presence of the TMEM106B risk allele was also found to reduce the age of onset by approximately 13 years compared with those without it,24 although this association was not confirmed in a recent GWAS study of GRN-related FTD.25 In the latter study, another variant that leads to increased expression of the GFRA2 gene was also found to be associated with increased risk for GRN-related FTD.25 The same major allele of TMEM106B associated with sporadic and GRN-related FTD also conferred an increased risk for C9orf72-related FTD and FTD-MND, but not MND.26 Interestingly, the T allele purported to be associated with shorter duration of disease in GRN-related FTD was associated with later age of onset and age of death in C9orf72-related FTD.27
Despite a better understanding of the pathophysiology and underlying genetic risk factors/modifiers of these disorders, evidence-based pharmacological interventions directed at mitigating their burden on patients are scarce. Currently, no intervention has been shown to alter the evolution of FTD spectrum disorders, and only a handful of small studies have demonstrated symptomatic benefits of pharmacological interventions.28 Designing interventional studies for these disorders is particularly challenging as a result of their low prevalence in the general population, insidious onset, and in many cases aggressive course.3 9 Moreover, in the absence of widely accepted sensitive and specific diagnostic biomarkers, the various clinical phenotypes, genetic and pathological heterogeneity, and the overlapping features of these disorders add to the complexity of designing valid clinical trials. Indeed, while the predictive value of established international consensus diagnostic criteria for these disorders continues to improve with every iteration,29–32 FTD spectrum disorders are frequently clinically misdiagnosed.33 Furthermore, with various cognitive, neuropsychiatric and motor manifestations, patients and caregivers’ needs are numerous and priorities difficult to establish. Finally, clinical heterogeneity makes it challenging for the development of meaningful clinical outcome measures that are sensitive to change across all of the diverse observed symptoms.
This article has several purposes. First, we present a critical review of the previously published randomised controlled trials (RCTs) of pharmacological interventions for FTD spectrum disorders wherein we (1) describe the populations studied; (2) examine and analyse the design, methodology and intervention applied in each trial; and (3) synthesise all the various outcomes of interest investigated, as well as the endpoints measured to date. Second, we present recommendations for designing future clinical trials in FTD spectrum disorders based on precision-medicine approaches to address the identified gaps and limitations of previous clinical studies.
Methods
Study design
A predesigned strategy was used for the literature search, study selection, data extraction and data synthesis. We adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.34 The protocol was registered prospectively on PROSPERO, where it can be retrieved and reviewed (trial registration number: CRD42018091194).
Search strategy and selection criteria
We performed a systematic review of the literature by using the databases MEDLINE, EMBASE and PsycINFO in order to identify RCTs of pharmacological interventions for the treatment of FTD spectrum disorders, more specifically behavioural and language variants of FTD (bvFTD, svPPA and nfvPPA), PSP syndrome and CBS. Although ALS shares some common molecular and genetic substrates with FTD disorders, and therefore often overlaps clinically with them, we chose to exclude trials on this condition as the design and methodology of ALS trials have already been critically reviewed previously.35 We used keywords including variations on ‘frontotemporal dementia’ and ‘clinical trial’ (see full search strategy in online supplementary material 2). We conducted our searches to retrieve articles from inception of databases up to 1 January 2018, without restrictions on language. We manually searched the reference lists of relevant reports for additional citations to supplement our electronic search. Two reviewers (PD and QDN) performed the literature research in parallel and independently. The reviewers met and selected articles to be included in the present study.
Supplemental material
Studies were included if they (1) investigated the effects of a pharmacological intervention; (2) were carried out in a population with an FTD spectrum disorder; (3) and were randomised and controlled. Studies were excluded if they (1) were observational/longitudinal studies or (2) represented only post-hoc analysis of previously published trials. Since the main purpose of our review was to identify limitations and gaps in previously published clinical trials, our selection process favoured inclusiveness. When disagreement arose between reviewers on studies to be included in the qualitative review, a third reviewer (MM) resolved the discrepancy.
Data extraction, quality assessment and statistical analysis
Data were abstracted in duplicate, concomitantly and independently by two trained investigators using a standard data abstraction form. Data pertaining to the study’s design (eg, condition studied, number of arms, type of intervention, comparator, eligibility criteria, primary and secondary endpoint measurements), the population (eg, size of groups, mean age and SD, percentage of women), the trial’s main conclusions, as well as the journal and date of publication were collected. Study quality was assessed with the Cochrane instrument to assess the risk of bias.36 When disagreements arose in coding, resolution was obtained through consensus. We performed descriptive statistics using IBM SPSS Statistics V.24.0.37
Results
Search results and study characteristics
A total of 947 abstracts were identified, of which 75 citations were reviewed at the full-text stage (see online supplementary figure S1 for the PRISMA flow chart). Of the articles reviewed in their entirety, 52 did not meet the inclusion criteria and were excluded. The 23 remaining articles were included in the present qualitative synthesis (table 1).38–60 Risk of bias in the included studies was perceived as low (see online supplementary data). From 1998 to 2016, a total of 1362 participants (44% female) with an FTD spectrum disorder were randomised to clinical trials. BvFTD and PSP syndrome were the most studied clinical conditions with 12 trials (52%) and 11 trials (48%) published, respectively. Three trials (13%) included participants with different subtypes of PPA. No interventional trial involving participants with CBS was identified. Of note, a large unpublished clinical trial of leuco-methylthioninium was also identified and involved 220 participants with a diagnosis of bvFTD. [S61] The largest trial in size for PSP syndrome also included participants with multiple system atrophy (MSA), with 363 participants with PSP syndrome (47%) and 404 with MSA (53%).50 Most trials were small in size, with only three trials (13%) having randomised more than 100 participants.
Supplemental material
Supplemental material
Randomised controlled trials of pharmacological interventions in FTD spectrum disorders
Several different drugs and regimens have been investigated as potential symptomatic therapies, with acetylcholinesterase inhibitors (donepezil, galantamine and rivastigmine) being the most frequently investigated drugs, followed by antidepressants (citalopram and paroxetine) and oxytocin, a neuropeptide (see online supplementary table S1). Investigations for potential disease-modifying agents were mainly performed in PSP syndrome and included coenzyme Q10,40 51 davunetide,46 rasagiline,39 riluzole,50 sodium valproate38 and tideglusib.44 45 A cross-over design was used in nine trials (39%), specifically for six symptomatic drug trials and three disease-modifying drug trials. The duration of interventions was shorter than 3 months for more than half of the trials (52%).
Supplemental material
Eligibility criteria in clinical trials
A summary of the main eligibility criteria for enrolment in these clinical trials is provided in table 2. For studies carried out in bvFTD, seven trials (58%) reported age requirements for inclusion, with a minimum age for inclusion ranging from 30 to 60, and the maximum age for inclusion ranging from 65 to 80. Eight trials (67%) explicitly excluded participants with advanced disease, defined by the presence of significant cognitive impairment. However, only four trials reported a specific threshold score for exclusion, which was based on the Mini-Mental State Examination (MMSE). Confirmatory abnormality on brain imaging, mainly frontotemporal atrophy, was the only biomarker requirement retrieved in the included clinical trials and was explicitly required for enrolment in seven of the bvFTD trials (58%). The scales used to assess severity of brain atrophy on imaging were not explicitly reported in the published articles, while the unpublished clinical trial in bvFTD [S61] enrolled participants with evidence of frontal and/or temporal lobe atrophy on MRI at Kipps level 2 or greater. [S62] None of the trials in bvFTD reported subsequent pathological confirmation following clinical diagnosis.
Eligibility and clinical diagnostic criteria of FTD spectrum disorders used in randomised controlled trials
For studies carried out in PPA, all three trials (100%) reported the use of age requirements, all excluding participants aged 80 years and older. Participants with advanced disease, defined by significant cognitive impairment on cognitive screening tests, were also excluded from these trials, with two trials (66%) reporting a specific MMSE threshold score for exclusion. The clinical diagnosis of enrolled PPA cases was supported by the presence of neuroimaging abnormalities, such as frontotemporal atrophy or hypoperfusion. The scales used to assess brain atrophy or hypoperfusion on imaging were not explicitly reported. None of the trials reported subsequent pathological confirmation following clinical diagnosis.
For studies conducted in PSP syndrome, all trials except one explicitly reported the use of age requirements for inclusion of participants. Six trials (55%) excluded participants with significant cognitive impairment, with four trials (36%) using a specific MMSE threshold score. Only two trials (18%) explicitly reported the use of neuroimaging for corroboration of clinical diagnosis. Finally, two trials (18%) mentioned subsequent pathological confirmation of the clinical diagnosis for a portion of the included participants.
Outcomes of interest and endpoint measures in clinical trials
Several different main outcomes of interest related to cognitive, language, neuropsychiatric and motor manifestations have been investigated in these clinical trials (table 1). Similarly, the effects of investigational drugs on the progression of symptoms were assessed through the use of various different scales and tools. The various scales and tools that have been used in these studies are reported in online supplementary table S2.
Supplemental material
All clinical trials in bvFTD but one focused on the treatment of neuropsychiatric symptoms, with the Neuropsychiatric Inventory [S63] and Frontal Behavioural Inventory [S64] being the most commonly used tools. The other trial53 investigated specifically the effect of methylphenidate on decision-making behaviour. Several scales and tools in regard to social cognition have also been used, including the Reading the Mind in the Eye Test, [S65] the Interpersonal Reactivity Index, [S66] and the Facial Expression Recognition Task. [S67] There were a variety of outcomes in clinical trials in PPA, including management of neuropsychiatric symptoms and palliation of language difficulties. All clinical trials in PSP syndrome except one explored the effects of investigational drugs on motor symptoms with the use of motor scales, with the Progressive Supranuclear Palsy Rating Scale (PSPRS) and the Unified Parkinson’s Disease Rating Scale being the most commonly used tools. One trial59 investigated specifically the effect of physostigmine on swallowing abilities. In regard to patient-centred outcomes, three trials (25%) in bvFTD and two trials (66%) in PPA reported measurements related to these, including tolerability of drug and impact on functional independency. In PSP syndrome, nine trials (82%) reported patient-centred outcomes, such as quality of life, autonomy and tolerability of treatment. Finally, only three trials (13%), all conducted in bvFTD, reported caregiver-related outcomes, such as caregiver burden.
Discussion
Heterogeneity in clinical trials
This systematic review of RCTs of pharmacological therapies for FTD spectrum disorders, with a focus on methodology, highlights some of the current challenges in designing and conducting clinical trials in these conditions. The significant heterogeneity in design and methodology of the identified clinical trials reflects the complexity of these syndromes and their underlying pathologies. Participants with different clinical phenotypes have been enrolled in these studies using diverse eligibility criteria based on the clinical diagnosis, age at baseline, as well as the presence or absence of certain cognitive deficits. Numerous drugs with different pharmacodynamic and pharmacokinetic properties have been investigated as potential disease-modifying interventions or symptomatic treatments of diverse symptoms and deficits (online supplementary table S1). Similarly, numerous tools and scales with different psychometric properties have been used to measure the effects of these investigational drugs (online supplementary table S2).
Limitations and challenges
Several limitations have been identified in these clinical trials, some of which could explain their negative results. First, clinical diagnosis for enrolment was not always detailed or clearly reported in the studies. This could be partly explained by the fact that the terminology has significantly evolved over the past decade as our understanding and conceptualisation of these disorders have improved. As well, inclusion of participants with concomitant MND was only reported explicitly in one study.47 Second, the use of biomarkers for corroboration of clinical diagnosis, or for exclusion of other neurocognitive disorders, was limited to the presence or absence of typical brain imaging abnormalities, mainly cerebral atrophy on CT scans and/or MRI, hypometabolism on FDG-positron-emission tomography (PET) scans, or hypoperfusion on single-photon emission CT scans in frontotemporal regions. With the notable exception of the Kipps scale used in one study [S61], the specific tools to assess brain imaging abnormalities were rarely reported in the retrieved studies. None of the trials explicitly reported the use of, for instance, cerebrospinal fluid analysis (CSF) or genetic testing for assessing the eligibility of participants. However, it must be stated that most of these clinical trials predated the discovery of genetic and other potential biomarkers.
Current international consensus clinical diagnostic criteria have limitations, as demonstrated in studies on neurocognitive disorders such as AD, Parkinson’s disease dementia and bvFTD. [S68–S71] Clinical diagnoses do not always match the final pathology results, with reported inaccurate diagnosis of AD pathology ranging from 19% to 45% in one cohort study. [S68] While recent iterations of clinical diagnostic criteria for FTD spectrum disorders have increased their sensitivities, such as Rascovsky’s criteria improving from previous diagnostic criteria of bvFTD (86% compared with 53% sensitivity),30 current criteria tend to have higher specificities than sensitivities.29–32 Thus, patients with some classical features of FTD may not meet the full criteria for diagnosis [S72] or can meet more than one criterion across different syndromes. [S71] Furthermore, multiple comorbid neurodegenerative and non-neurodegenerative findings may be present at autopsy. [S73–S77] For instance, α-synucleinopathies (24.9%), tauopathies (23.2%), TDP-43 proteinopathy (13.3%) and vascular lesions (48.9%) were identified in the brains of elderly individuals with and without dementia in a large community-based autopsy series. [S73] Hence, definitions of neurodegenerative disorders based exclusively on clinical signs and symptoms may not fully grasp the heterogeneity of underlying pathologies. Third, clinical trials in bvFTD where brain atrophy on imaging was not required for participants’ eligibility may have unintentionally randomised phenocopy cases. These slowly progressive and sometimes non-progressive cases, which fulfil the neuropsychological and neuropsychiatric criteria for bvFTD, appear to be neuropathologically distinct from other forms of FTD at autopsy, with the notable exception of C9orf72 mutation cases. [S78] Previously enrolled participants of RCTs may not have had the expected underlying pathology; thus, the drug being tested might have been off target. Fourth, most of the retrieved studies were early-phase clinical trials aimed at assessing safety and tolerability, hence their short durations and small cohort sizes. These could preclude the detection of a treatment effect especially relating to disease modification, which would need potentially a longer observation period. With the exception of a few multicentre trials that were able to recruit larger cohorts, most clinical trials identified were small in size and may have lacked statistical power. Single-centre trials often had a cross-over design, while multicentre trials had a classic placebo-controlled, parallel-group design. Fifth, previous RCTs of pharmacological interventions may have been attempted too late in the course of the disease, potentially missing the therapeutic window of opportunity. Finally, selected tools and clinical scales used to measure treatment effect may not have been sensitive enough to capture significant changes in symptoms or disease progression as they may not cover effectively all of the various clinical manifestations of the FTD spectrum or fail to take into account the functional-anatomical specificity of the frontal regions.
Addressing limitations and moving forward
In the context of complex clinical manifestations and pathological heterogeneity, uniformity and standardisation in future clinical trials for FTD spectrum disorders are needed. Precision-medicine approaches, where interventions are designed by considering the patient’s specific clinical syndrome as well as disease profile in regard to its underlying molecular and genetic signatures, offer opportunities to address some of the current challenges. The following elaborated recommendations for future clinical trials in the FTD spectrum disorders should be considered (see table 3).
Recommendations for future clinical trials in FTD spectrum disorders
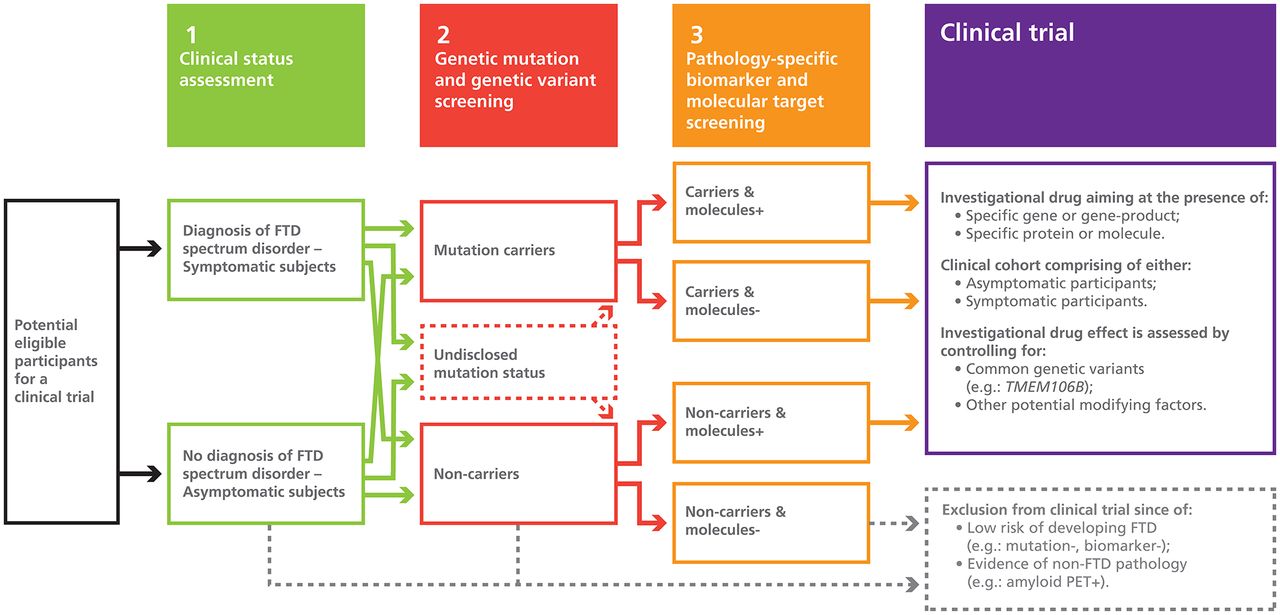
The diagnoses of participants enrolled in clinical trials and their supportive findings should be clearly defined and reported. Initial clinical diagnoses should be prospectively reassessed as participants’ clinical presentation may change over time. [S71, S79–S81] Additionally, subsequent pathological confirmation of participants’ diagnoses should be attempted whenever possible. This could be facilitated by systematically discussing brain donation with every eligible participant before enrolment in a clinical trial. FTD spectrum disorders have a wide range of clinical manifestations, which frequently overlap with each other.3 33 Therefore, investigations allowing exclusion of other disorders that may mimic FTD spectrum disorders, such as other neurodegenerative disorders (eg, AD), psychiatric disorders and vascular disease, should be rigorously performed. [8 14 33 S75,S76] As well, phenocopies of bvFTD should also be identified as they can also contribute to pathological misclassification in clinical trials. [S82] Although it may be difficult to accomplish this before randomisation, clinical suspicion should increase in the context of participants with non-progressive conditions. FTD spectrum disorders are biologically heterogeneous, involving several different pathological inclusion proteinopathies, variants and subtypes, and cerebral topographies, which should all be considered in clinical trials.11 14 [S83–S87] Although there are still no widely accepted biomarkers that are both sensitive and specific, some molecular and genetic findings could potentially be used as eligibility criteria for clinical trials in FTD spectrum disorders. Possible examples include plasma progranulin levels, where decreased levels can predict the presence of GRN mutations, [S88–S92] and plasma and CSF neurofilament light chain protein levels, where levels could reflect disease severity. [S93–S96] In the (hopefully) near future, a tau-ligand PET scan could help increase accuracy of clinical diagnoses, where FTLD cases due to TDP-43 proteinopathy would be PET-negative and cases due to tauopathy would be PET-positive. [S97–S100] Considering the important contribution of genetic mutations and variants to these disorders, genetic testing should be an integral part of the selection process of participants. [S101–S104] A known genetic profile is informative of the expected natural progression of the underlying disorder. For instance, C9orf72 promoter hypermethylation is associated with prolonged disease duration in expansion carriers. [S105–S106] Although uncommon, the possibility of co-occurrence of genetic mutations should also be considered, such as C9orf72 repeat expansion carriers also harbouring GRN or MAPT gene mutations. [S107] Moreover, molecular and genetic factors could potentially represent interesting targets for future drugs, such as the open-label trial of nimodipine in progranulin deficiency, [S108] as well as markers of pharmacological response or adverse effects, similar to previously identified genetic drug response markers in Parkinson’s disease. [S109] With improvement of target engagement, specifying whether the diagnosis of a participant is based solely on clinical signs or in conjunction with molecular, genetic or histopathological findings is essential as these markers significantly improve the prediction of the correct underlying pathological process and hence the presence of the pharmacological target (see figure 1).

Selection process of participants for FTD spectrum disorders clinical trials based on precision-medicine approaches. A graphical representation of the proposed selection and triage process of potential eligible participants for future clinical trials in FTD spectrum disorders based on precision-medicine approaches. First, subjects are assessed clinically to determine the presence of a clinical syndrome according to the international consensus diagnostic criteria (eg, bvFTD). Then, symptomatic and asymptomatic subjects undergo genetic testing to identify mutation carriers (eg, C9orf72, GRN, MAPT, VCP and TARDP), as well as genetic variants (eg, APOE epsilon 4 allele, HLA, MAPT H1 haplotype and TMEM106B T allele) that may modify age of onset or increase the risk of a specific pathological substrate. In the circumstance that potential eligible participants would not want to be informed of their mutation status, they could still be able to enrol in clinical trials. Finally, subjects undergo further testing to identify the presence of FTLD-specific biomarkers and molecular targets (eg, progranulin plasma level and CSF tau level). This selection process permits the exclusion of subjects with a low risk of developing FTLD and those with non-FTLD pathology, such as Alzheimer’s disease. Clinical trials can then be conducted in a population with a well-characterised disease where the investigational drug’s target is present. bvFTD, behavioural variant FTD; CSF, cerebrospinal fluid; FTD, frontotemporal dementia; FTLD, frontotemporal lobar degeneration; PET, positron-emission tomography.
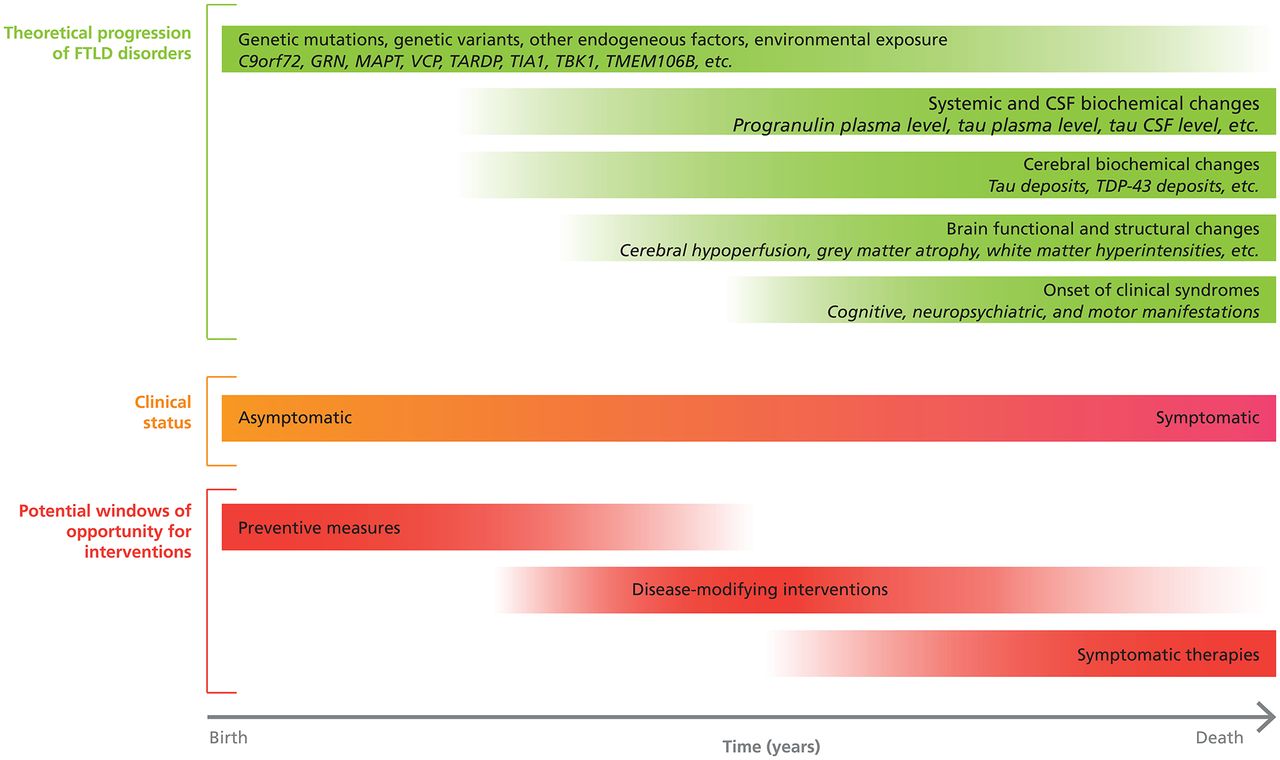
Different clinical trials should involve participants at diverse stages of the disease, including presymptomatic stages. Similar to AD, FTD spectrum disorders progress over years before clinical manifestations and brain changes such as hypometabolism, hypoperfusion and atrophy become apparent (see figure 2). [S110, S111] Although penetrance of known mutations may vary considerably according to several factors (eg, TMEM106B genotype), future clinical trials should investigate potential disease-modifying interventions in high-risk asymptomatic individuals, who are mutation carriers. Identifying and recruiting presymptomatic individuals in studies may be difficult but is feasible, as demonstrated by the Genetic Frontotemporal Dementia Initiative (GENFI), [S111] the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS), [S112] and the Advancing Research and Treatment for FTLD (ARTFL) studies. [S113]
{kind=link}
{kind=link}
Windows of opportunities for pharmacological interventions in FTD spectrum disorders according to disease progression. A graphical representation of the theoretical progression of FTD spectrum disorders over time and potential windows of opportunity for pharmacological interventions. CSF, cerebrospinal fluid; FTD, frontotemporal dementia; FTLD, frontotemporal lobar degeneration; TDP-43, TAR DNA-binding protein 43.
Since FTD spectrum disorders have a low prevalence and incidence in the general population compared with AD,1 2 innovative methodologies and study designs should be sought. Statistically meaningful results require enrolment of a sufficient number of participants. Therefore, international initiatives and collaborations, such as the Neuroprotection and Natural History in Parkinson Plus Syndromes Study50 and ‘clinical trial ready’ cohorts such as GENFI, [S111] LEFFTDS, [S112] and ARTFL [S113] are essential to test new clinical questions and to establish biomarkers that can be used as outcome measures. Initiating, implementing and maintaining international networks come with challenges and obstacles, such as additional financial costs, cultural and language differences, and data transmission issues, just to name a few. But these collaborations are highly valuable in the long run. The Dominantly Inherited Alzheimer Network-Trials Unit represents an example of a successful international clinical trial in autosomal dominant AD. [S114] International research registries and networks facilitate recruitment of potential research participants and promote alliances between healthcare providers and researchers.
Conducting adaptive clinical trials with Bayesian models and platform trials, such as the Glioblastoma Adaptive Global Innovative Learning Environment, [S115] are interesting avenues to investigate multiple prospective pharmacological interventions aimed at specific target points in a short amount of time. [S116,S117] Furthermore, in the absence of effective treatments, pharmacological and non-pharmacological interventions, such as exercise, should be coinvestigated in parallel and in combination in order to maximise chances of identifying interventions with symptomatic benefits. Successful drug-tailoring for rare diseases is possible, such as it was recently done in spinal muscular atrophy with the development of nusinersen, an antisense oligonucleotide drug. [S118,S119] Similar to therapies in oncology and microbiology, administration of several drugs with different pharmacodynamic properties in combination may be necessary in order to have a disease-modifying effect in FTD spectrum disorders.
Exclusion criteria should be carefully planned so as not to exclude certain subgroups of individuals. Historically, women [S120,S121] and the elderly [S122–S125] have been under-represented in clinical trials. There is evidence of sex differences in the clinical manifestations of genetic mutations in FTD, which could translate into differential treatment effects. [S126] Specifically, there appears to be a higher prevalence of female patients with GRN-related FTD [S126], indicating that future clinical trials in GRN-related FTD should adjust for potential sex effects. Although exclusion of participants with advanced age can be justified by the increased prevalence of comorbid cerebral pathology, [S73,S76,S127–S129] the higher risk of adverse effects of investigational drugs, [S130,S131] and different progression rates of diseases, [S132,S133] differential treatment effects should be explored by subgroup analyses. Older age at onset may also increase the likelihood of an FTD case being sporadic where underlying pathologies and risk factors are different from those in early-onset cases, which may be more likely to have an identifiable genetic mutation. Similarly, exclusion of participants with cognitive impairment should be clearly justified. Cognitive deficits are nearly universal during the course of FTD spectrum disorders, and the ideal screening tool and threshold score for inclusion/exclusion remain to be determined. The MMSE is not the most sensitive nor specific screening tool for the assessment of cognitive deficits in these disorders. [S134,S135] As well, the MMSE may not be discriminative enough to help in determining the stage of these disorders early in their course. For instance, language impairment affects the assessment of other cognitive domains, and the use of a specific threshold score for inclusion may inappropriately exclude participants with aphasia from clinical trials, despite them having little or no other important cognitive or behavioural impairment. [S135]
Researchers should prioritise the investigation of pharmacological interventions aimed at patient-centred and caregiver-centred outcomes. There is currently an unmet need for effective symptomatic therapies at all stages of these disorders. Patients, families and caregivers should be involved as research partners in the decision process in order to identify and prioritise goals to pursue in future clinical trials. The Association for Frontotemporal Degeneration, the FTD Disorders Registry and the FTD Treatment Study Group are all encouraging these and other partnerships. As well, studies should clearly define and report whether the outcomes of interest of the investigated treatment relate to prevention, symptomatic relief and/or disease modification. Clinical trials with longer observation periods should be undertaken in order to capture disease-modifying effects of investigational drugs.
FTD spectrum disorders have various cognitive, neuropsychiatric and motor manifestations, which may require different pharmacological interventions to treat them, and consequently necessitate different assessment tools and scales to measure the effects of the interventions. Tools specifically designed for FTD spectrum disorders, which take into consideration the various manifestations of these disorders (ie, global composite measures), should be prioritised, such as the Clinical Dementia Rating Scale-FTLD [S136–S138] and the PSPRS. [S139,S140] Tools assessing disease severity in specific domains, such as frontal system functions, should be used in conjunction. [S141] As well, it should be acknowledged that some clinical manifestations may interfere with the assessment of several other cognitive, neuropsychiatric and motor impairments, affecting interpretation of results. Functional scales taking this fact into consideration should also be prioritised, such as the CBD-Functional Rating Scale. [S142] Multidimensional patient-reported and caregiver-reported measures should likewise be integrated in future clinical trials. An interesting avenue includes the Goal Attainment Scaling instrument, which is a personalised outcome measure where patients and caregivers set the treatment goals. [S143] Another alternative approach to identifying a treatment effect is to capture change in symptoms and functions using a variety of different rating instruments previously used, but then to identify a composite clinical effect through the use of eigenfunctions and multifactor dimensionality reduction approaches applied to the comprehensive data set collected. In addition, change in neuroimaging measures over time, especially in the case of presymptomatic prevention trials, should also be explored as a potential outcome measure to consider in conjunction with clinical measures. These include reduction in the rate of atrophy on MRI and reduction in tau burden on PET (once a viable tracer has been validated). Furthermore, increases in plasma and/or CSF progranulin levels for GRN-related FTD may also be a possible treatment goal, although there has not been a good correlation observed between disease measures and progranulin levels to date. In addition, the arrival of new technologies to assist with the assessment and monitoring of individuals with cognitive and functional deficits, such as wearable devices, is an exciting moment in dementia research and could offer novel ways to capture the effects of clinical interventions in the near future.
Finally, with our increased understanding of rare, causative mutations for genetic FTD and common genetic variation that increases risk for sporadic FTD or that modifies its course in genetically confirmed or sporadic cases, clinical trial design should consider stratified designs based on the presence of an autosomal dominantly inherited mutation, and/or inclusion of genetic modifiers in the analysis as a covariate to account for variability in the course of FTD. This will allow better control for factors that vary substantially from person to person in FTD, such as age at onset or rate of disease progression.
Conclusion
Clinical trial research in FTD spectrum disorders is in its infancy. Individuals afflicted with these neurodegenerative disorders have numerous unmet needs. Development of new pharmacological interventions specifically designed for these individuals is essential as no effective disease-modifying treatments or evidence-based symptomatic therapies have been identified. Critical examination of previously published RCTs revealed potential explanations for their negative results as well as opportunities to improve future endeavours. We hope these recommendations, which are based on patient-centred and precision-medicine approaches, will help to steer clinical trials in FTD spectrum disorders in a productive direction.
Additional references can be found in the online supplementary material 1.
Supplemental material
Glossary
Frontotemporal dementia (FTD)—refers to the clinical diagnoses of behavioural variant FTD and primary progressive aphasia (ie, semantic and non-fluent variants), which are mainly based on clinical manifestations and, sometimes, supported by the presence of characteristic cerebral hypometabolism, hypoperfusion or atrophy on brain imaging.
Frontotemporal dementia spectrum—refers to the clinical diagnoses of behavioural variant FTD and primary progressive aphasia (ie, semantic and non-fluent variants), and to corticobasal syndrome, progressive supranuclear palsy syndrome and amyotrophic lateral sclerosis.
Frontotemporal lobar degeneration (FTLD)—refers to neuropathological diagnoses where brain pathological examination reveals frontal and/or temporal lobe atrophy on macroscopy, and tau, TDP-43 or FUS immunoreactive inclusions on microscopy. For FTLD due to tau, subtypes include Pick’s disease, corticobasal degeneration, progressive supranuclear palsy, FTDP-17, globular glial tauopathy and argyrophilic grain disease.
Patient-centred outcome—refers to outcomes that are meaningful to patients, such as quality of life and autonomy.
Precision-medicine—refers to interventions individually tailored on the basis of a patient’s environmental exposure, lifestyle factors, genes, proteins, proteomics and imaging.
Acknowledgments
PD and QDN would like to thank Fondation du Centre Hospitalier de l’Université de Montréal for supporting their postdoctoral training in cognitive neurology and epidemiology, respectively. PD would like to thank Sylvain G Bélisle for his help with the creation of the figures.
References
Footnotes
Contributors PD and MM were involved in the preparation of project conception. PD, QDN and MM were involved in the review’s execution. PD created tables and figures, performed the statistical analyses, and prepared the initial draft of the paper with MM. Manuscript review and critique were performed by all authors. The final version was read and approved by all authors, with PD and MM incorporating their additional comments.
Funding This study was supported by operating grants from the Canadian Institutes of Health Research (MOP 327387), and the Weston Brain Institute to MM.
Competing interests PD and QDN have nothing to disclose. SEB and MM are supported by the Department of Medicine (Sunnybrook Health Sciences Centre and the University of Toronto), the Sunnybrook Foundation, the Hurvitz Brain Sciences Research Program and the Sunnybrook Research Institute. MM also receives support as co-lead of the Ontario Neurodegenerative Disease Research Initiative funded by the Ontario Brain Institute. SEB also receives support as the executive director of the Toronto Dementia Research Alliance. SEB does contract research for Eli Lilly, Biogen, GE Healthcare, Genentech, Novartis, Optina and Roche; received peer-reviewed funding from CIHR, CPSR, Brain Canada, ADDF, NIA, NIH, Leducq Foundation, OBI and Weston Brain Institute; and received speaker fees from Novartis and Eli Lilly. MM receives peer-reviewed research support from the Canadian Institutes of Health Research, Weston Brain Institute, Washington University, Parkinson Society Canada, Alzheimer’s Drug Discovery Foundation, Brain Canada, Canadian Consortium on Neurodegeneration in Aging and Ontario Brain Institute. MM has also served as an advisor to Bioscape Medical Imaging CRO, Novartis, Ionis Pharmaceuticals, Arkuda Therapeutics and UCB; received honoraria from Novartis; received royalties from Henry Stewart Talks; received an investigator-initiated research grant from Teva; and received contract research support from Roche, Novartis and Axovant. NH is supported by the Canadian Consortium on Neurodegeneration in Aging and peer-reviewed grants from the Alzheimer Society of Canada (grant 15-17), Alzheimer’s Drug Discovery Foundation (grant 20140503), Canadian Institutes of Health Research, National Institute on Aging of the National Institutes of Health (grant R01AG046543), and the Heart and Stroke Foundation (grant NA 7220), in addition to research contracts funded by Roche, Axovant and Lundbeck Canada. NH received consultation fees from Astellas, Lilly, Merck, Pfizer and Mediti. AEL has served as an advisor for AbbVie, Acorda, Biogen, Bristol-Myers Squibb, Janssen, Sun Pharma, Kallyope, Merck, Paladin and Corticobasal Degeneration Solutions; received honoraria from Sun Pharma, Medichem, Medtronic, AbbVie and Sunovion; received grants from Brain Canada, Canadian Institutes of Health Research, Corticobasal Degeneration Solutions, Edmond J Safra Philanthropic Foundation, Michael J Fox Foundation, the Ontario Brain Institute, National Parkinson Foundation, Parkinson Society Canada and W Garfield Weston Foundation; received publishing royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins Press and Cambridge University Press. HHF receives peer-reviewed grant funding support from the Canadian Consortium on Neurodegeneration in Aging (CIHR 137794), National Institute on Aging (U19AG010483) (2UF1Ago32438-07), CIHR and Weston Brain Institute (Grant # 363926), and Brain Canada (IMPACT AD study), and other grant funding from Toyama Pharmaceutical, Biohaven Pharmaceuticals and Probiodrug. He has either current or past service agreements through UC San Diego with Banner Health, Roche Genentech, Arkuda Therapeutics, Samus, Samumed, Eisai, Merck and TauRx, and has received travel expenses from Axon Neurosciences, Alion Pharmaceuticals and Probiodrug. All other authors declare no competing interests.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.