Article Text

Abstract
Objective To evaluate the efficacy and safety of intramuscular ultra-high-dose methylcobalamin in patients with amyotrophic lateral sclerosis (ALS).
Methods 373 patients with ALS (El Escorial definite or probable; laboratory-supported probable; duration ≤36 months) were randomly assigned to placebo, 25 mg or 50 mg of methylcobalamin groups. The primary endpoints were the time interval to primary events (death or full ventilation support) and changes in the Revised ALS Functional Rating Scale (ALSFRS-R) score from baseline to week 182. Efficacy was also evaluated using post-hoc analyses in patients diagnosed early (entered ≤12 months after symptom onset).
Results No significant differences were detected in either primary endpoint (minimal p value=0.087). However, post-hoc analyses of methylcobalamin-treated patients diagnosed and entered early (≤12 months’ duration) showed longer time intervals to the primary event (p<0.025) and less decreases in the ALSFRS-R score (p<0.025) than the placebo group. The incidence of treatment-related adverse events was similar and low in all groups.
Conclusion Although ultra-high-dose methylcobalamin did not show significant efficacy in the whole cohort, this treatment may prolong survival and retard symptomatic progression without major side effects if started early.
Trial registration number NCT00444613.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is an intractable neurodegenerative disease characterised by motor neuron degeneration typically presenting with muscle weakness and atrophy.1 Respiratory failure due to muscle weakness is the major cause of death. Without mechanical ventilation support, patients succumb to this disease within 3–6 years from its onset.
The widely used drug for ALS, riluzole, provides modest prolongation of survival (2–3 months), but no beneficial effects were shown on muscle strength and little on bulbar function.2 Moreover, safety concerns, such as liver dysfunction, exist.2 Edaravone has been approved for retarding the clinical deterioration of ALS, but its effect on the survival is unknown.3
The deficiency in vitamin B12 is associated with central nervous system lesions including subacute combined degeneration of the cord, indicating an important function of B12 in the spinal cord and the brain. Methylcobalamin, an active vitamin B12, used in Japan to treat peripheral neuropathy and megaloblastic anaemia, is a potential candidate for ALS treatment. It functions as a coenzyme for homocysteine remethylation as a methyl donor, and inhibits neuronal degeneration by decreasing levels of homocysteine, the accumulation of which contributes to neuronal degeneration in patients with ALS.4 5 It also activates extracellular signal-regulated kinases 1 and 2 and Akt to induce neurite outgrowth and prolong neuronal survival.6 Cyanates/cyanide conjugates of B12 are not acting as methyl donor and have not been proven to show these effects.
Preclinical studies have reported that methylcobalamin protects neurons against glutamate neurotoxicity7 8 and promotes nerve regeneration.9 It has also been shown that intraperitoneal ultra-high dosage inhibits disease progression in a wobbler mouse.10 Oral administration in high dose would be ineffective because of the limited availability of the gastric intrinsic factor for its absorption. Clinical studies have demonstrated the efficacy of intramuscular ultra-high-dose methylcobalamin on compound muscle action potentials.11 Moreover, a small-sized study has demonstrated that, if started early in the disease course, ultra-high-dose methylcobalamin prolongs ventilation-free survival.12
Based on these results, we conducted a long-term phase II/III clinical trial to evaluate the efficacy and safety of intramuscular ultra-high-dose methylcobalamin in Japanese patients with ALS.
Methods
This multicentre, randomised, double-blind, placebo-controlled clinical trial was conducted from December 2006 to March 2014 at 51 sites in Japan.
Patients
Patients satisfying the following inclusion criteria were eligible: outpatients aged 20 years or older; clinically definite, clinically probable, or clinically probable, laboratory-supported ALS diagnosis according to the revised El Escorial criteria (Airlie House criteria)13; duration from the symptom onset ≤3 years; stage 1 or 2 of the Japanese ALS severity classification14 (scores range from 1 to 5: 1 denotes no difficulty in daily living and working; 2, ability to live or work unaided; 3, requirement of assistance in daily living due to incapability of managing social life; 4, requirement of constant assistance in all aspects of daily living; and 5, bedridden status requiring a life support system); and Revised ALS Functional Rating Scale (ALSFRS-R) score15 decrease by 1–3 points during the 12-week observation period. The definition of onset was the initial time that the patient recognised weakness or any other motor symptoms other than twitching or cramping of muscles. The key exclusion criteria were tracheostomy or previous use of non-invasive ventilation, per cent-predicted forced vital capacity (%FVC) ≤60%, multiple conduction blocks, new start or change in the dose or administration of riluzole after the observation period began, or serious cardiovascular, renal, hepatic disease or any haematological changes suggestive of B12 deficiency.
This report follows the Consolidated Standards of Reporting Trials guidelines.
Randomisation and treatment
The patients were centrally randomised to the placebo or 25 mg or 50 mg methylcobalamin groups using the order of registration with a minimisation algorithm to balance the following factors: onset type (bulbar or upper or lower motor neuron onset), riluzole coadministration, ALSFRS-R score before study enrolment, and the change in this score during the observation period.
Allocated drugs were intramuscularly administered twice per week starting from the end of the observation period (12 weeks) and continued for 182 weeks in a manner that the patients and their caregivers could not see the formulation colour (the active ingredients in methylcobalamin colour the formulation red). Changes in riluzole administration were not allowed. Edaravone was not used in any of the subjects.
Outcome measures
The primary endpoints were the time to primary events and the change in ALSFRS-R score from baseline to week 182. The primary events were defined as death by any cause or invasive or non-invasive ventilation support ≥22 hours per day due to ALS progression. On the occurrence of a primary event, treatment was discontinued. ALSFRS-R quantitatively evaluates the progression of disability by measuring respiratory function and physical ability in daily living.
The secondary endpoints included muscle strength assessed using the manual muscle test (Medical Research Council Scale), physical functional status measured with the Norris Scale,16 respiratory function assessed using %FVC, grip strength, and the quality of life evaluated using the ALS Assessment Questionnaire-40.17
The primary and secondary efficacy endpoints were evaluated using post-hoc analyses in a subgroup of patients diagnosed early (at screening ≤12 months after symptom onset) based on previous studies.11 12
Drug safety was evaluated on the incidence of adverse events and the results of laboratory tests, vital signs and an ECG. Events due to the progression of ALS were not counted as adverse events; however, all deaths were counted as adverse events regardless of cause.
All assessments, except for ECG, were conducted on weeks 0, 4 and 16, and at 12-week intervals thereafter to week 172, and on week 182. For patients who discontinued therapy due to the primary event, the last assessment was conducted within 4 weeks of the day the event occurred. Different investigators were responsible for drug administration, as well as efficacy, ALSFRS-R and safety assessments, to maintain blindness throughout the study, because the active ingredients in methylcobalamin colour urine red.
Statistical analysis
The sample size was originally set at 200 primary events in 300 patients (100 per group) for 130 weeks to detect a significant difference in the HR for the time to primary event between the groups at a one-sided significance level of 1%, with a power of 90%, based on an estimated HR of the primary events of 0.5–0.6 and effect size in the ALSFRS-R analysis of 0.3–0.4. However, the sample size and study duration were revised while maintaining blindness to 360 patients (120 per group) and 182 weeks because of a low rate of primary events.
Two interim analyses by an independent data monitoring committee were performed to assess safety and futility.
The efficacy analyses were conducted using a population analysis for those patients who received methylcobalamin and had evaluable primary endpoint data based on the intention-to-treat principle. This was called the full analysis set. The safety analyses were made on a safety data set composed of those patients who were evaluated for safety. Missing data from patients who discontinued methylcobalamin after a primary event were imputed with the final evaluation data after discontinuation.
Methylcobalamin efficacy and dose–response relationships were simultaneously evaluated using contrast coefficients to compare placebo with 50 mg and placebo with all methylcobalamin groups combined, respectively. The time interval to the primary event was compared among groups using log-rank scores, and the changes in the ALSFRS-R score and secondary endpoint measures were evaluated using the Wilcoxon score (patients who died or whose data after a primary event were not collected within 28 days from the event were ranked worst). The p values for the primary test were adjusted for multiplicity, with the statistical analysis plan described in online supplementary e-appendix. In addition, the Cox proportional hazards model with backward elimination for the placebo group was used with variables including the interval between symptom onset and diagnosis (≤12 months, >12 months), gender, %FVC (<90%, ≥90%) and several other variables to explore prognostic factors for events post hoc. Analyses were performed using SAS V.9.3 software.
Supplemental material
Results
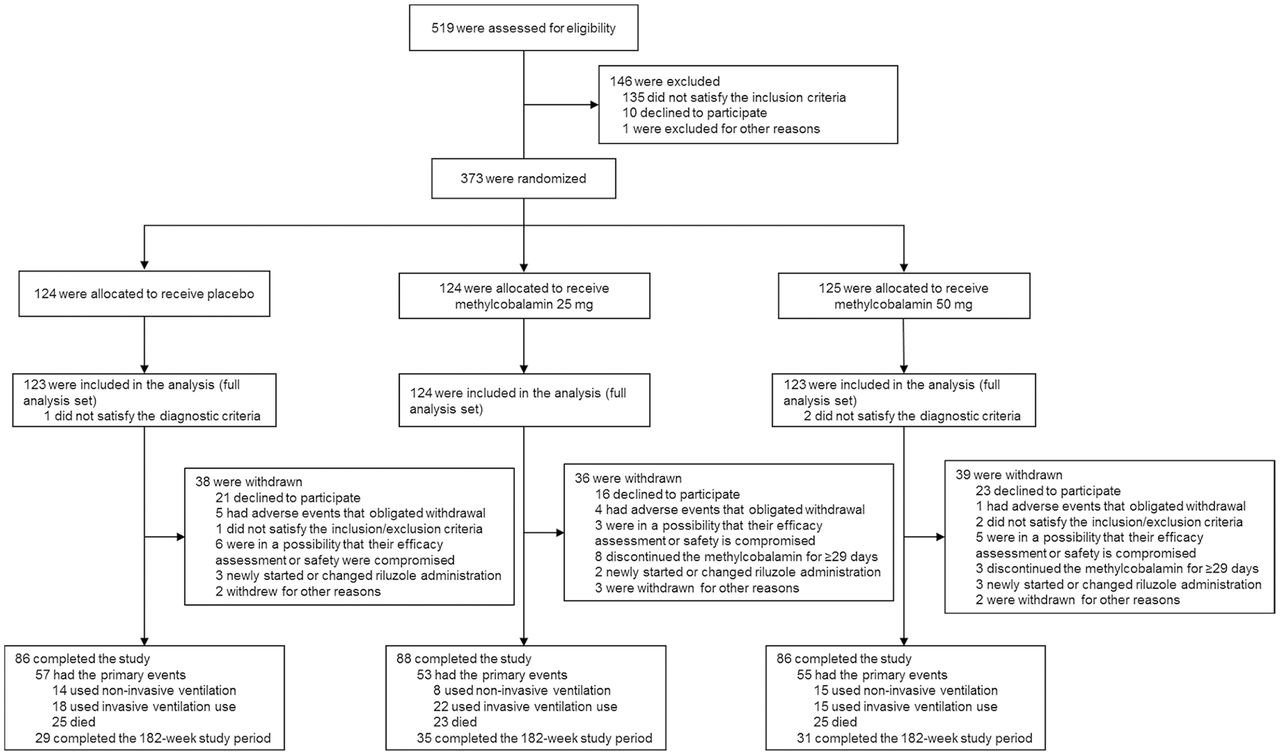
A total of 373 patients were enrolled and randomly assigned to placebo or 25 mg or 50 mg methylcobalamin groups (124, 124 and 125 patients, respectively; figure 1). Exclusion of 3 patients (1 and 2 patients in the placebo and 50 mg groups, respectively) for not satisfying the diagnostic criteria yielded 370 patients. The study was completed by 260 patients, with 113 patients withdrawn because they declined to participate.
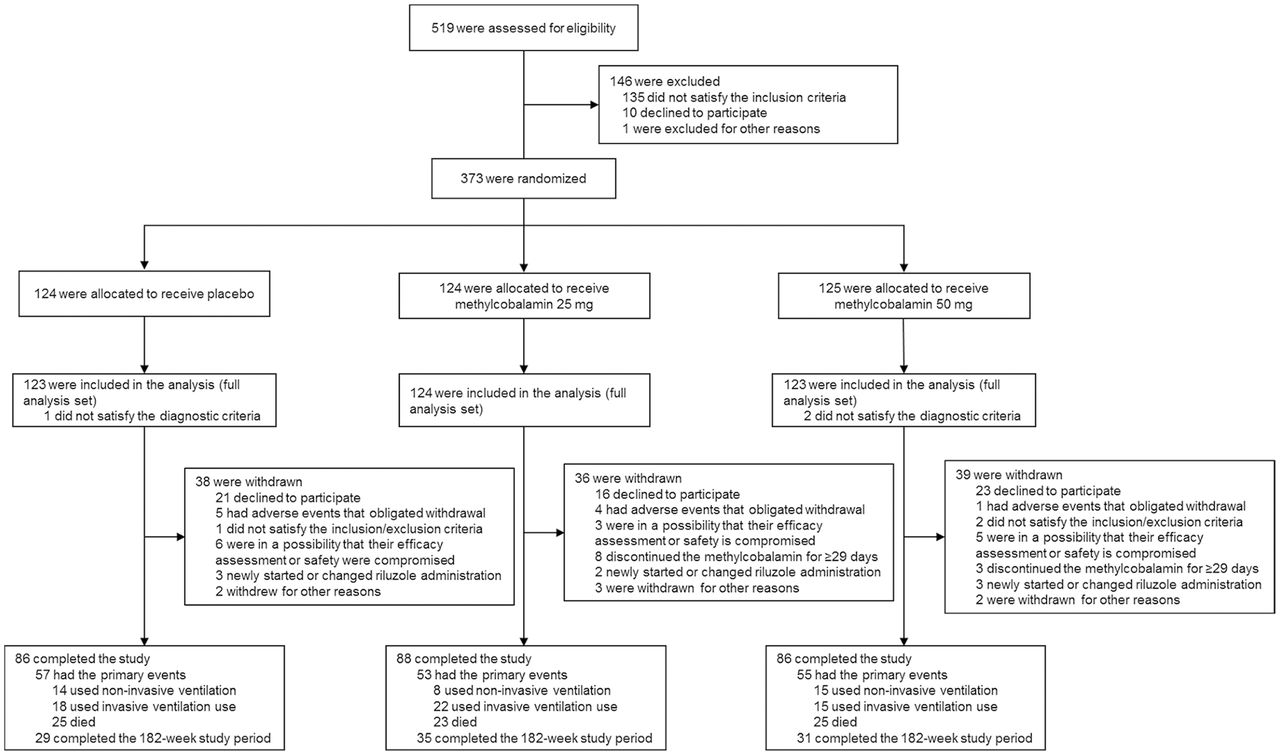
Patient flow.
The baseline demographic and disease characteristics were similar among the groups, without significant differences (table 1). Approximately half the patients were diagnosed as having clinically probable ALS (46.2%) and had upper motor neuron-onset ALS (49.5%). Most of the patients (89.7%) were being treated with riluzole at the screening. The number of patients with diabetes was 53 (16 in placebo, 18 in 25 mg and 19 in 50 mg groups), 6 of whom received metformin, which could potentially affect B12 levels. There were however no changes in haematological data in this study.
Baseline characteristics of study participants
Efficacy
Significant differences were not detected for either primary endpoint; the minimal crude p value was 0.09 for the change in the ALSFRS-R score, and its adjusted value was 0.19 (table 2). The time to the primary event was slightly prolonged in the active treatment groups (HR [95% CI]: 0.83 [0.58 to 1.20] for 25 mg and 0.92 [0.65 to 1.32] for 50 mg methylcobalamin groups). The median time to the primary event was 880 for placebo, 1147 for 25 mg and 954 days for 50 mg methylcobalamin groups (figure 2A). The median change in the ALSFRS-R score from baseline to week 182 decreased in relation to the allocated dose: −24.0 for placebo, −22.0 for 25 mg and −21.0 for 50 mg methylcobalamin groups (figure 2B).

{kind=link}
{kind=link}
Primary efficacy endpoints in all patients (A, B) and the subgroup of patients diagnosed early (≤12 months after symptom onset) (C, D). ALSFRS-R, Revised Amyotrophic Lateral Sclerosis Functional Rating Scale.
Primary efficacy endpoints analysed in two patient populations
For the secondary endpoints, the median change in manual muscle test and the Norris Scale scores from baseline to week 182 decreased in a dose-dependent manner, although this decrease was not significantly different among the groups (online supplementary table e-1).
Supplemental material
Patients diagnosed early (duration ≤12 months)
The post-hoc analysis of the subgroup of patients diagnosed early (diagnosed with ALS ≤12 months after symptom onset) demonstrated a significant dose–response-dependent prolongation in time to the primary event (HR [95% CI]: 0.64 [0.38 to 1.09] for 25 mg [p=0.01] and 0.50 [0.27 to 0.93] for 50 mg [p=0.01] methylcobalamin groups). The median time to the primary event (95% CI) was 570 (465 to 720) days for placebo, 1087 (564 to –) days for 25 mg, and 1197 (609 to –) days for 50 mg methylcobalamin groups (table 2, figure 2C).
The change in the ALSFRS-R score also decreased in a dose-dependent manner (the p value for 25 mg was 0.01 and was 0.003 for 50 mg methylcobalamin compared with placebo; figure 2D). To confirm the validity of the results, the time-related changes in the efficacies on the primary event and on ALSFRS-R scores were analysed; methylcobalamin exhibited efficacy or a trend towards efficacy on primary events in patients diagnosed ≤12 months after symptom onset. Additionally, efficacy or a trend towards efficacy on ALSFRS-R scores was frequently observed in the first 24 months after symptom onset (online supplementary tables e-2 and e-3).
Among the secondary endpoints, a dose–response inhibition in worsening was shown in the Norris Scale score (p values: 0.008 for 25 mg and 0.005 for 50 mg) and %FVC (p values of 0.004 for 25 mg and <0.001 for 50 mg) (online supplementary table e-4).
Patients with other poor prognostic factors
Applying the Cox proportional hazards model with backward elimination to data in the placebo group determined the following poor prognostic factors: diagnostic interval >12 months, being male, %FVC being <90% and being without riluzole (online supplementary table e-5). Methylcobalamin at both 25 mg and 50 mg tended to reduce the HR in men and %FVC <90% (HR, 0.76–0.77; online supplementary table e-6). The decreased ALSFRS-R score (95% CI) in the 50 mg group was 4.3 (0.7 to 7.9; p=0.095) for men and 4.5 (0.9 to 8.1; p=0.020) for %FVC <90% (online supplementary table e-7).
Safety
Adverse events were reported by more than 97% of patients in each group. Treatment-related adverse events were reported with a similar incidence of 4.1% (5/123), 7.3% (9/124) and 5.7% (7/123) in the placebo, 25 mg and 50 mg methylcobalamin groups, respectively. The incidence of serious adverse events was also similar in the placebo, 25 mg and 50 mg methylcobalamin groups: 64.2%, 62.1% and 65.0%, respectively. Of the six patients who died of causes other than ALS progression, the cause of one death in the 50 mg methylcobalamin group was due to cardiac arrest following myocardial infarction or arrhythmia, and was considered unrelated to the medication based on the patient’s history. There were no clinically significant changes in the results of laboratory tests, vital signs or ECGs among groups. Statistical details are available on request.
Classification of evidence
The research aims of this study were to evaluate the efficacy and safety of long-term ultra-high-dose methylcobalamin (25 mg and 50 mg) in Japanese patients with ALS and the efficacy in patients whose ALS was diagnosed early. Methylcobalamin was not found to be significantly superior to placebo in the whole cohort. However, in patients diagnosed early (≤12 months after symptom onset), this study provides post-hoc class II evidence that ultra-high-dose methylcobalamin prolongs time to death or ventilation support (HR [95% CI]: 0.64 [0.38 to 1.09] for 25 mg group and 0.50 [0.27 to 0.93] for 50 mg group; p=0.01 for placebo vs both methylcobalamin groups combined) and decreased ALSFRS-R scores (p=0.003 for 50 mg and p=0.01 for all methylcobalamin groups) in a dose-responsive manner. The incidence of treatment-related adverse events was similar and low in all groups.
Discussion
This long-term study evaluated the efficacy and safety of high-dose methylcobalamin (25 mg and 50 mg administered intramuscularly twice per week) in patients with ALS using the survival (or being fully bound to respirator) as the primary event. Because of the time and expenses incurred, it is becoming more and more difficult to conduct large-scale, long-term studies assessing the survival such as in the present study.
Although the superiority of methylcobalamin over placebo in terms of either the time to primary events or the change in ALSFRS-R score was not confirmed when data from all participants were analysed, a post-hoc analysis using only the subgroup of patients diagnosed early (diagnosed ≤12 months after symptom onset) demonstrated the efficacy of methylcobalamin. This subgroup suggested dose–response relationships for both survival prolongation and functional measures.
Despite the lack of a statistically significant difference compared with placebo as a whole, deterioration in ALSFRS-R, Manual Muscle Testing (MMT) and Norris scales scores tended to be less pronounced with the higher dose of methylcobalamin. These findings warrant a pivotal phase III clinical trial of ultra-high-dose methylcobalamin exploring its effect on ALSFRS-R for a shorter period (16 weeks), recruiting patients with less than 12 months’ duration after onset (Clinical Trial of Ultra-high Dose Methylcobalamin for ALS (JETALS), ClinicalTrials.gov NCT03548311).
The length of time between the first ALS symptom and the initial clinic visit correlated with patient survival, likely because those with rapid progression tend to be captured early by the current diagnostic criteria.18 Those participants with less time between symptom onset and initial clinical visit exhibited shorter survival times. In the present study, the percentage of patients in the placebo group who experienced a primary event (95% CI) was 75.6% (60.1% to 90.1%) for those with a duration of ALS ≤12 months and 51.8% (38.8% to 64.7%) for those >12 months. The median change in the ALSFRS-R scores from baseline to week 182 was −26.5 for those ≤12 months and −21.0 for those >12 months after symptom onset.
Besides those with early diagnosis (ie, rapid disease progression), a similar trend towards poor prognosis regarding time to primary events (or survival) was found in the other two subgroups: being male and having %FVC <90% (online supplementary table e-6). Interestingly, in these three subgroups, methylcobalamin also tended to show a positive influence in the survival and in other endpoints (ALSFRS-R, Norris Scale score and %FVC; online supplementary table e-7). These results could reinforce the efficacy of methylcobalamin in ALS. Provided that findings in favour of methylcobalamin were obtained only in these subgroups and not in the whole cohort, it may be difficult to evaluate the efficacy of a therapeutic drug in patients with slow and variable progression of the disease.
Alternatively, methylcobalamin may be more efficacious when treatment is started at an early stage of ALS. A recent clinical trial of erythropoietin recruiting patients up to 18 months after onset showed a tendency for longer survival and less decrease of ALSFRS-R score up to 6 months.19 Baumann and colleagues20 demonstrated that the half-life of lower motor neurons is approximately 1 year and that these motor neurons decay exponentially in ALS. This means that the number of lower motor neurons is already halved at 1 year after symptom onset. Therefore, in modifying the progression of ALS, a therapeutic agent started late in the progression has only a fraction of the normal lower motor neuron population remaining, with a small number of lower motor neurons sustaining a large number of muscle fibres.
As previously suggested21 the reason so many clinical trials may fail despite the promising results of animal studies is partly due to the late treatment start in humans compared with that in animal models. The El Escorial criteria up to the clinically probable, laboratory-supported level may not be sensitive enough to detect patients with ALS in the early stage of the disease for participation in clinical trials, in contrast to the detection criteria used in animal studies.
In clinical settings, the average delay from symptom onset to an ALS diagnosis is approximately 1 year, and a delay of ≤12 months accounts for about 40% of patients.21–23 The newly developed Awaji criteria,24 incorporated into the El Escorial criteria, may shorten the delay to 9 months.25–28 In this study, methylcobalamin showed prominent prolongation of survival with slower functional decline in patients diagnosed early (≤12 months after symptom onset). Currently, approximately half of the ALS population could benefit from methylcobalamin treatment using the revised El Escorial diagnostic criteria; however, if the use of the Awaji criteria becomes the standard practice, most patients could benefit from this therapy, if the promise is fulfilled in the currently ongoing JETALS study, which uses the Awaji criteria for entry for the first time. The inconvenience of intramuscular injections may be overcome by allowing injections by patients or their caregivers, which is currently employed in the JETALS study.
Further limitations of this trial should be noted. First, the strict criteria for study inclusion may have excluded some patients with ALS, which has heterogeneous pathogeneses. Second, although post-hoc analysis identified only one subgroup of patients, additional factors may influence the efficacy and safety profile of methylcobalamin. Third, we did not examine higher doses (>50 mg) for dose finding, and it is possible that these mega-doses might have even better outcome. These potential factors may warrant future analyses in other study cohorts such as JETALS.
In conclusion, ultra-high-dose methylcobalamin was not found to be significantly superior to placebo. However, ultra-high-dose methylcobalamin therapy may improve the prognosis of patients with ALS if administered early in the disease course. Therapeutic agents that failed in the previous clinical trials could be reanalysed for potential efficacy in ALS, taking into account the duration of the disease at the start of therapy. Criteria enabling earlier diagnosis and a change in the physician’s attitude towards offering an early diagnosis and treatment should yield better future outcomes for the patients than ever.
Acknowledgments
We thank all patients for their participation and the site staff for their contributions. Contributors (trial conduct supervision by site): Ichiro Yabe (Hokkaido University Hospital), Jun Kawamata (Sapporo Medical University Hospital), Hiroto Takada (National Hospital Organization Aomori Hospital), Keiji Chida (National Hospital Organization Iwate Hospital), Masashi Aoki (Tohoku University Hospital), Hiroaki Ito (National Hospital Organization Miyagi Hospital), Masashiro Sugawara (Akita University Hospital), Muneshige Tobita (National Hospital Organization Yonezawa Hospital), Yoshihiro Sugiura (Fukushima Medical University Hospital), Mitsuya Morita (Jichi Medical University Hospital), Yukio Fujita (Gunma University Hospital), Yuichi Maruki (Saitama Neuropsychiatric Institute), Katsuhisa Ogata (National Hospital Organization Higashi Saitama Hospital), Kimihito Arai (National Hospital Organization Chiba-East-Hospital), Hidehiro Mizusawa (Tokyo Medical and Dental University Hospital), Masashi Takanashi (Juntendo University Hospital), Yasuo Iwasaki (Toho University Omori Medical Center), Takashi Sakamoto (National Center Hospital, National Center of Neurology and Psychiatry), Mieko Ogino (The Kitasato Institute, Kitasato University East Hospital), Kazuko Hasegawa (National Hospital Organization Sagamihara Hospital), Ichiro Imafuku (Japan Labour Health and Welfare Organization, Yokohama Rosai Hospital), Kazuhiro Muramatsu (Saiseikai Yokohama-shi Tobu Hospital), Ryoko Koike (National Hospital Organization Nishi-Niigata Chuo Hospital), Yousuke Yonemochi (National Hospital Organization Niigata Hospital), Kiyonobu Komai (National Hospital Organization Iou Hospital), Hiroyuki Yahikozawa (Nagano Red Cross Hospital), Koichi Mizoguchi (National Hospital Organization, National Epilepsy Center, Shizuoka Institute of Epilepsy and Neurological Disorders), Tsuyoshi Uchiyama (Seirei Social Welfare Community, Seirei Hamamatsu General Hospital), Naoki Atsuta (Nagoya University Hospital), Kazuya Nokura (Fujita Health University Banbuntane Hotokukai Hospital), Akira Taniguchi (Mie University Hospital), Kengo Maeda (Shiga University of Medical Science Hospital), Hideyuki Sawada (National Hospital Organization Utano Hospital), Toshiki Mizuno (University Hospital Kyoto Prefectural, University of Medicine), Harutoshi Fujimura (National Hospital Organization Toneyama Hospital), Kenya Murata (Wakayama Medical University Hospital), Yuetsu Ihara (National Hospital Organization Minami-Okayama Medical Center), Kouichi Noda (National Hospital Organization Higashihiroshima Medical Center), Masanori Hiji (Mifukai Vihara Hananosato Hospital), Chigusa Watanabe (National Hospital Organization Hiroshima-Nishi Medical Center), Takeo Yoshimura (Shakaihoken Shimonoseki Kohsei Hospital), Hiromasa Fukuba (National Hospital Organization Yanai Medical Center), Ryuji Kaji (Tokushima University Hospital), Shuji Hashiguchi (National Hospital Organization Tokushima Hospital), Masahiro Nomoto (Ehime University Hospital), Yasushi Osaki (Kochi Medical School Hospital), Hiroyuki Nakazawa (Nankoku Hospital), Hitoshi Kikuchi (Murakami Karindoh Hospital), Tomoaki Yuhi (Hospital of University of Occupational and Environmental Health, Japan), Tomoko Narita (National Hospital Organization Nagasaki Kawatana Medical Center) and Masahito Suehara (National Hospital Organization Okinawa Hospital). Draft manuscript editing was provided by Clinical Study Support (Nagoya, Japan) under contract with Eisai. Statistical analysis was conducted by Yasuo Ohashi, PhD, Chuo University, Department of Integrated Science and Engineering for Sustainable Society, and Takao Takase, MSc, Eisai.
References
Footnotes
Contributions RK designed the study, interpreted the data and drafted the manuscript. TI designed the study. YI designed the study. KO designed the study. MN designed the study. YO designed the study and performed the statistical analysis. TT designed the study, performed the statistical analysis, interpreted the data and drafted the manuscript. TH interpreted the data and drafted the manuscript. HS designed the study, interpreted the data and drafted the manuscript. KT designed the study and interpreted the data. SK designed the study and interpreted the data.
Funding This study was funded by Eisai.
Competing interests RK received grants from Eisai during the conduct of the study and has a patent on the Method of treating amyotrophic lateral sclerosis (US 20130344081 A1 licensed). TI received grants from Eisai during the conduct of the study. YI reports no disclosures. KO reports no disclosures. MN reports no disclosures. YO received personal fees from Statcom, Sanofi, Eisai, Chugai, Taiho, Shionogi and Kowa, and non-financial support from Yakult Honsha and Takeda outside the submitted work. TT is employed by Eisai. TH is employed by Eisai. HS is employed by Eisai. KT received grants from Eisai during the conduct of the study. SK received grants from Eisai during the conduct of the study.
Patient consent for publication Not required.
Ethics approval This study is conducted in accordance with the principles of the Declaration of Helsinki. The protocol was approved by the institutional review board at each centre. All eligible patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.