Article Text

Abstract
Objective To evaluate the long-term safety and efficacy of deutetrabenazine in patients with tardive dyskinesia (TD).
Method Patients with TD who completed the 12 week, phase 3, placebo-controlled trials were eligible to enter this open-label, single-arm study. The open-label study consisted of a 6 week dose-escalation phase and a long-term maintenance phase (clinic visits at Weeks 4, 6 and 15, and every 13 weeks until Week 106). Patients began deutetrabenazine at 12 mg/day, titrating up to a dose that was tolerable and provided adequate dyskinesia control, based on investigator judgement, with a maximum allowed dose of 48 mg/day (36 mg/day for patients taking strong cytochrome P450 2D6 (CYP2D6) inhibitors). Safety measures included incidence of adverse events (AEs) and scales used to monitor parkinsonism, akathisia/restlessness, anxiety, depression, suicidality and somnolence/sedation. Efficacy endpoints included the change in Abnormal Involuntary Movement Scale (AIMS) score (items 1 to 7) from baseline and the proportion of patients rated as ‘Much Improved’ or ‘Very Much Improved’ on the Clinical Global Impression of Change.
Results A total of 343 patients enrolled in the extension study, and there were 331 patient-years of exposure in this analysis. The exposure-adjusted incidence rates of AEs with long-term treatment were comparable to or lower than those observed in the phase 3 trials. The mean (SE) change in AIMS score was –4.9 (0.4) at Week 54 (n = 146), – 6.3 (0.7) at Week 80 (n = 66) and –5.1 (2.0) at Week 106 (n = 8).
Conclusions Overall, long-term treatment with deutetrabenazine was efficacious, safe, and well tolerated in patients with TD.
Trial registration number NCT02198794.
- tardive dyskinesia
- deutetrabenazine
- movement disorders
- vmat2 inhibitor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Tardive dyskinesia (TD) is a movement disorder caused by dopamine-receptor antagonists (DRAs), such as typical and atypical antipsychotics and antiemetics, including metoclopramide and prochlorperazine.1–7 The uncontrolled abnormal movements of TD can be socially stigmatising, disabling and worsen quality of life.8–10 Clinicians may initially attempt to manage TD by lowering the dose of or discontinuing the causative agent, but this may not be possible, as it could exacerbate symptoms of the existing primary psychiatric disease or worsen TD symptoms.1–3 5
Deutetrabenazine (chemical name: RR, SS)−1, 3, 4, 6, 7, 11b-hexahydro-9, 10-di(methoxy-d3)−3-(2-methylpropyl)−2H-benzo(a)quinolizin-2-one) is a novel, highly selective vesicular monoamine transporter 2 inhibitor that contains deuterium, a naturally occurring, non-toxic form of hydrogen.11–13 Deutetrabenazine has been approved in the USA for the treatment of chorea in Huntington disease and tardive dyskinesia.14 In two phase 3, randomised, double-blind, placebo-controlled, 12 week trials (ARM-TD15 and AIM-TD16), patients with TD who were treated with deutetrabenazine showed significant improvements in TD symptoms. In ARM-TD (N=117), patients receiving deutetrabenazine (titrated to a recommended dose of 48 mg/day for most patients) significantly improved TD symptoms compared with placebo based on the change from baseline to Week 12 in blinded central video-rated Abnormal Involuntary Movement Scale (AIMS) scores (–3.0 vs –1.6, p=0.019).15 At the end of the titration period, the mean total daily dosage of deutetrabenazine was 38.8 (SD: 7.92) mg/day, which remained stable until the end of the treatment period (38.3 (SD: 7.97) mg/day).15 In AIM-TD (N=298), patients receiving deutetrabenazine (titrated to 12, 24 or 36 mg/day) showed significant improvement in AIMS score for the 36 mg/day (–3.3 vs –1.4, p=0.001) and 24 mg/day (–3.2 vs –1.4, p=0.003) dosages compared with placebo.16 In both trials, there were low rates of overall adverse events (AEs) and discontinuations associated with deutetrabenazine. Importantly, deutetrabenazine treatment did not interfere with the use of concomitant DRAs for underlying psychiatric conditions.15 16
The purpose of this open-label extension of the ARM-TD and AIM-TD studies was to evaluate the safety, tolerability and efficacy of long-term maintenance therapy with deutetrabenazine. Open-label outcomes through Week 106 are reported in this analysis.
Method
Study design
This open-label, single-arm, long-term study of deutetrabenazine in patients with TD (ClinicalTrials.gov NCT02198794) was conducted at 76 centres in the USA and Europe. Patients with TD who successfully completed one of the phase 3 trials (ARM-TD and AIM-TD) were eligible to roll over into this open-label study. All patients were washed out from the phase 3 study drug (deutetrabenazine or placebo) for at least 1 week and began deutetrabenazine at 12 mg/day (6 mg twice daily), regardless of prior treatment. Deutetrabenazine dose was then increased weekly by 6 mg/day over the first 6 weeks of treatment (dose-escalation phase) until adequate dyskinesia control was achieved with good tolerability. All treatment regimens were administered twice daily, approximately 10 hours apart. The maximum total daily dosage was 48 mg/day, unless patients were taking strong CYP2D6 inhibitors (paroxetine, fluoxetine or bupropion), in which case maximum dose was 36 mg/day. Patients continued this established dose into the long-term treatment period. The data for this long-term analysis was 5 April, 2017.
Study participants
Study participants successfully completed either the ARM-TD or AIM-TD trial, defined as participation through Week 13; compliance with study drug and procedures, in the opinion of the investigator; and the absence of ongoing AEs that are serious or severe in intensity, or are expected to interfere with participation in this study. Patient inclusion and exclusion criteria have been previously described.15 16
Each patient, or their legally authorised representative, signed and dated an Institutional Review Board/Independent Ethics Committee-approved Informed Consent Form.
Blinding
This was an open-label study and was therefore not blinded.
Outcomes
A patient was considered compliant if the patient took 80% to 105% of the expected tablets throughout the overall treatment period (the number of deutetrabenazine tablets used: (tablets dispensed – tablets returned) divided by the expected number of tablets used).
Safety was assessed by: the raw incidence of AEs, clinical laboratory tests, vital signs, weight, electrocardiography (ECG) with machine reading of cardiac intervals, physical and neurological examinations and concomitant medication use. Exposure-adjusted incidence rates (EAIRs) were used to compare AE frequencies in this open-label extension study with those in short-term treatment in ARM-TD and AIM-TD. EAIRs were determined by adjusting the incidence of AEs by the duration of treatment exposure. EAIRs were calculated for patients in the following groups: patients given placebo in ARM-TD and AIM-TD (pooled group), patients treated with fixed doses of deutetrabenazine (12 mg/day, 24 mg/day and 36 mg/day) in the AIM-TD study, patients titrated with deutetrabenazine in the ARM-TD study and all patients receiving deutetrabenazine in this open-label extension study. Safety assessments were also made by the following rating scales: Unified Parkinson’s Disease Rating Scale (UPDRS) motor examination, Hospital Anxiety and Depression Scale (HADS), Barnes Akathisia Rating Scale (BARS), Columbia Suicide Severity Rating Scale (C-SSRS), Epworth Sleepiness Scale (ESS) and the Montreal Cognitive Assessment (MoCA).
Efficacy of deutetrabenazine was evaluated over the long-term treatment period, with 11 post-baseline clinic visits up to Week 106 (Weeks 2, 4, 6, 15, 28, 41, 54, 67, 80, 93 and 106). The change in total motor AIMS score (items 1 to 7, score range 0 to 28) from baseline to each visit was used to assess the severity of TD over time. In contrast to the phase 3 trials, which employed centralised video assessments, this analysis used local site ratings of the AIMS. The Clinical Global Impression of Change (CGIC) is a single-item questionnaire asking the investigator to assess the patient’s TD symptoms after starting treatment; similarly, the Patient Global Impression of Change (PGIC) asks the patient to assess their TD symptoms at specific visits after starting therapy. Both the CGIC and PGIC use a 7-point Likert scale ranging from ‘Very Much Worse’ to ‘Very Much Improved’. The proportion of patients ‘Much Improved’ or ‘Very Much Improved’ (treatment success) at each visit on both the CGIC and PGIC was another efficacy variable. Quality of life was assessed using a modified version of the Craniocervical Dystonia Questionnaire (mCDQ-24). Certain domains (stigma, emotional well-being, pain, activities of daily living and social/family life) were modified to focus on the impact of TD.
Statistical analyses
Safety analyses were performed in the safety population, which included all enrolled patients who received any study drug. Safety data compared the overall population to the phase 3 double-blind trail outcomes and were summarised using descriptive statistics.
Efficacy analyses were performed on the intent-to-treat (ITT) population, which included all enrolled patients, regardless of whether the patient received study drug. No inferential statistical analyses were done for efficacy endpoints; rather, descriptive statistics were used to summarise these endpoints at each visit.
Results
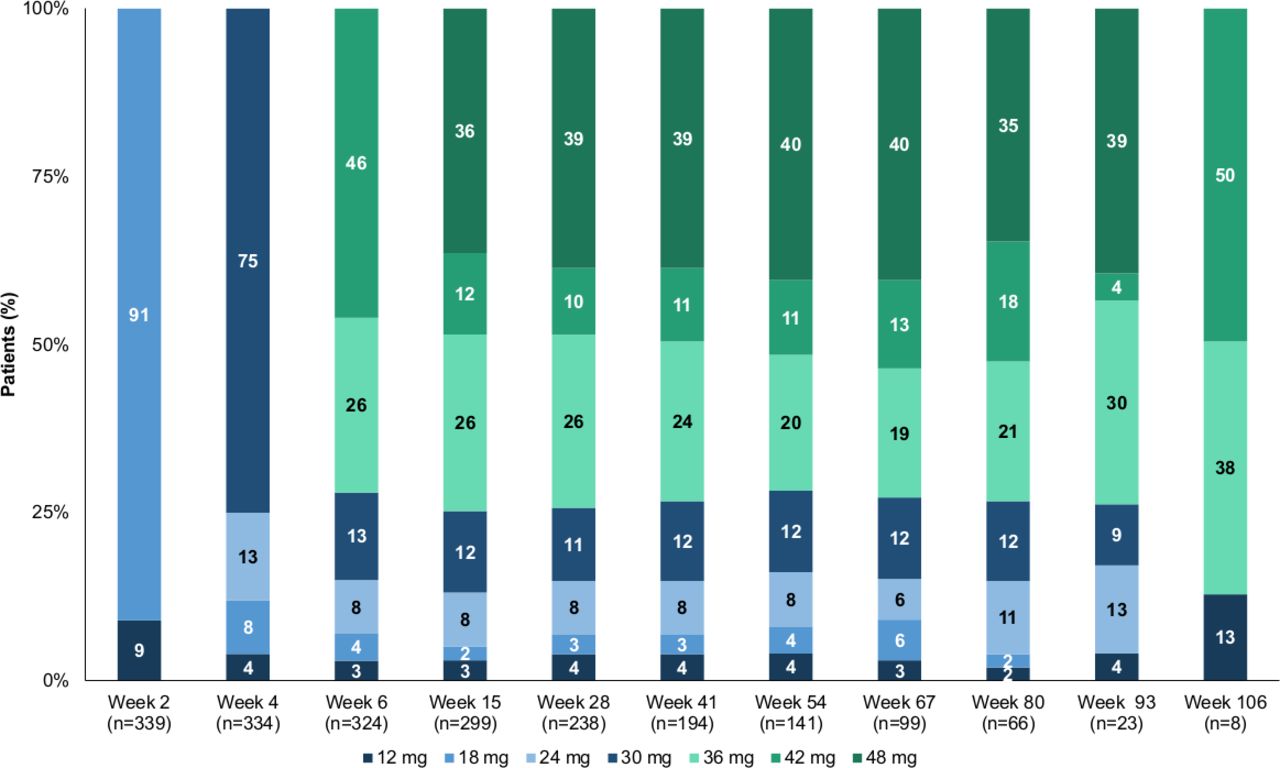
Of the 368 patients who successfully completed the phase 3 trials, 343 patients rolled over into this open-label extension study between 15 October, 2014, and the data cut-off date of 5 April, 2017. As of the cut-off, all eligible patients had either enrolled into this study or declined to participate. A total of 25 patients did not roll over, most commonly due to withdrawal of consent, not meeting selection criteria or the centre not participating in the open-label study. Of the 343 patients enrolled, 232 had received deutetrabenazine and 111 had received placebo in the phase 3 trials. As of the cut-off, 105 patients had discontinued the study, with patient withdrawal from the study as the most common reason, while 238 patients (69%) were ongoing in the study (figure 1). All patients that enrolled (ITT population, n=343) were treated with study drug and were included in the safety population. Baseline patient data from the phase 3 trials show similar demographics for age, sex, race and weight, based on study treatment (prior deutetrabenazine and prior placebo) (table 1). Mean (±SE overall treatment compliance was 90.2%±1.0%, with a compliance rate from ≥80% to 105% (105% compliance was defined as patients taking 5% more tablets than expected) in 77% (264/343) of patients. Mean total daily dose of deutetrabenazine for all patients was 38.1 mg (SE: 0.9) at Week 54, 38.6 mg (SE: 1.1) at Week 80 and 39.0 mg (SE: 4.4) at Week 106. At Week 80, the total daily dose level distribution was: 1/66 (2%) for 12 mg, 1/66 (2%) for 18 mg, 7/66 (11%) for 24 mg, 8/66 (12%) for 30 mg, 14/66 (21%) for 36 mg, 12/66 (18%) for 42 mg and 23/66 (35%) for 48 mg (figure 2). Mean duration of deutetrabenazine treatment was 352.9 days, with 76% (259/343) of patients receiving at least 54 weeks of treatment and 20% (69/343) of patients receiving at least 80 weeks of treatment.
Patient baseline characteristics by study treatment group

Patient disposition. Patients enrolled between 15 October, 2014, and 5 April, 2017 (data cut-off date). ITT, intent to treat.

Percentage of patients at each deutetrabenazine dose level over the long-term treatment period. The total daily dose level distribution for 12 mg, 18 mg, 24 mg, 30 mg, 36 mg, 42 mg and 48 mg through week 106 are presented. Highest dose levels (48 mg) were reached by week 80 in 35% of patients.
A total of 233 patients experienced an AE in the open-label extension, with an EAIR of 1.68, comparable to or lower than those seen with short-term, double-blind deutetrabenazine treatment (EAIR range 2.85 to 6.50) and placebo (EAIR 3.92) (table 2). The incidence rate of serious adverse events (SAEs) in the open-label extension (EAIR 0.15) was similar to those observed during short-term blinded deutetrabenazine treatment (EAIR range 0.13 to 0.39) and placebo (EAIR 0.33) (table 2). Three patients had SAEs considered possibly related to deutetrabenazine: intentional overdose/suicide attempt, exacerbation of mania and exacerbation of hypomania. The patient who experienced intentional overdose and suicide attempt had a history of depression, bipolar disorder, anxiety, suicide attempts and suicidal thoughts, and was on 18 mg/day of deutetrabenazine at the time of the events. Study discontinuations, dose reductions and dose suspensions due to AEs were also uncommon, and EAIRs were comparable to those seen with short-term deutetrabenazine treatment and placebo (table 2).
Exposure-adjusted incidence rate of patients with AEs in the overall treatment period
Most AEs were mild or moderate in severity, and the most common AEs included anxiety, somnolence and depression (table 2). Incidence rates during open-label extension were comparable to those observed with short-term deutetrabenazine and placebo treatment, indicating no evidence of cumulative toxicity or tolerability findings associated with long-term deutetrabenazine treatment (table 2). Of the 38 AEs leading to discontinuation (occurring in 26 participants), 16 events occurring in 11 participants were serious AEs. Of these, only two were adjudicated as possibly related to study drug. The incidence of AEs was not correlated with the CYP2D6 poor metaboliser genotype; however, this may be due to the low number of poor metabolisers in the study population (n=11).
Other safety measures, as assessed by rating scales, did not show meaningful changes during the long-term treatment period. There was no worsening of anxiety based on the HADS subscale, parkinsonism based on the UPDRS motor examination, akathisia and restlessness based on the BARS, somnolence and sedation based on the ESS or cognitive function based on the MoCA (online supplementary table 1). The depression measure worsened by a mean (SD) change of 0.7 (3.4) based on the HADS subscale (0 to 21-point scale, online supplementary table 1); however, due to a lack of comparator group, we are unable to perform statistical analyses other than descriptive statistics. EAIRs during the open-label extension for depression, anxiety, suicidality, akathisia and restlessness, somnolence and sedation and parkinsonism occurred at a similar frequency to those seen with short-term treatment with deutetrabenazine and placebo (table 2, online supplementary table 2).
Supplemental material
Based on the C-SSRS, suicidal ideation was reported in 16/342 patients (5%) and suicidal behaviour was reported in 2/342 patients (<1%) (online supplementary table 1). A total of seven patients (EAIR 0.02) patients reported suicidality AEs: intentional overdose, suicide attempt and suicidal ideation. Three had serious suicidality AEs. One patient experienced two serious suicidality AEs on the same day (intentional overdose and suicide attempt) that were possibly related to the study drug. Of the 16 participants with a reported C-SSRS during the study, only two had no prior medical history of depression or suicidal behaviour: One had a prior suicide attempt, eight had a history of bipolar disorder and eight had a history of depression. There were six deaths, three of which were considered unrelated to the study drug (septic shock, brainstem infarction and cardiac failure) and the other three were considered unlikely to be related to the study drug (cardiac arrest, ventricular tachycardia and cardiovascular insufficiency/respiratory failure). Importantly, a recent retroactive study of patients with Huntington disease (N=4091) found no association of tetrabenazine use with increased risk of depression or suicidality.17
There were no clinically meaningful changes in laboratory measurements (serum chemistry, haematology and urinalysis) over the long-term treatment period. Body weight showed small changes over the long-term treatment period, with a mean decrease of 0.2 kilograms (SE: 1.13) at Week 80. Vital signs and ECG parameters also did not show clinically meaningful changes over the treatment period. Post-baseline Fridericia-corrected QT interval (QTcF) abnormalities occurred as follows: QTcF >480 ms in 6/343 (2%) patients and QTcF >500 ms in 3/343 (<1%) patients.
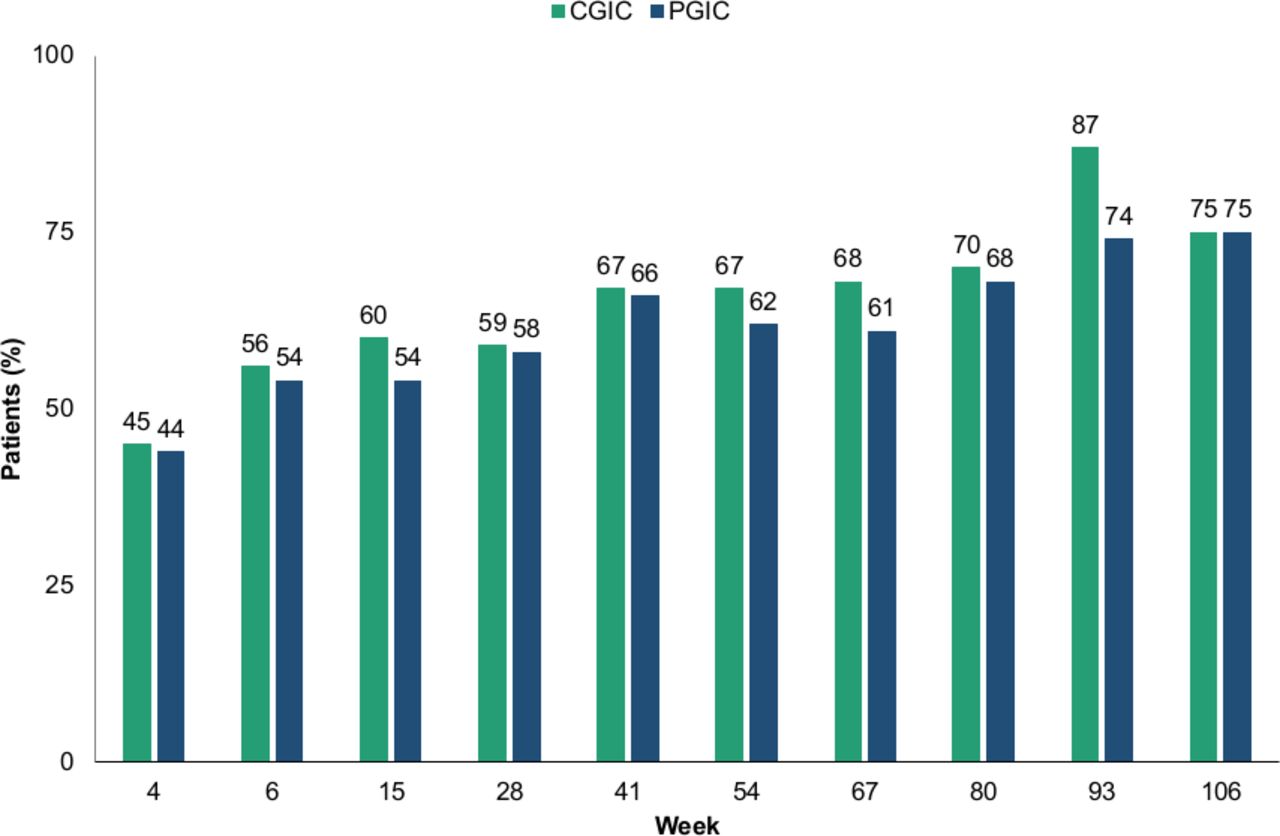
Site-rated efficacy outcomes showed improvements over the long-term treatment period. There was a gradual reduction from baseline in the mean AIMS score starting at Week 2 (mean change (SE): –1.7 (0.17)) and this trend continued through Week 80 (–6.3 (0.66)) (figure 3). After 6 weeks of deutetrabenazine treatment, 56% (184/327) of all patients were ‘Much Improved’ or ‘Very Much Improved’ as measured by the CGIC, and 70% (46/66) of patients who reached Week 80 of the study reported the same (figure 4).
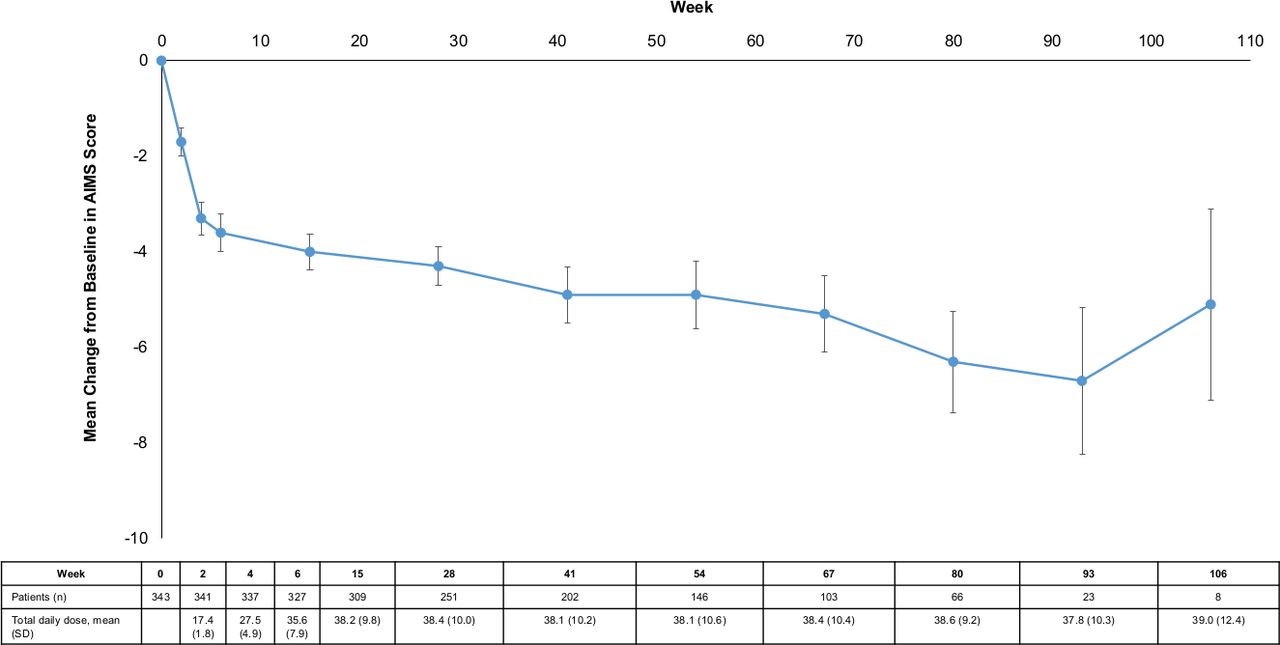
Mean change in AIMS score over the long-term treatment period. Site-rated AIMS score showed improvements over the long-term treatment period. There was a gradual reduction from baseline in the mean AIMS score starting at week 2 that continued through week 106. Error bars represent SE. AIMS, Abnormal Involuntary Movement Scale; SD, standard deviation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patients ‘Much Improved’ or ‘Very Much Improved’ on CGIC and PGIC over the long-term treatment period. Patients were ‘Much Improved’ or ‘Very Much Improved’ as measured by the CGIC and PGIC after 6 weeks of deutetrabenazine treatment, and continued to show improvements through week 106. CGIC, Clinical Global Impression of Change; PGIC, Patient Global Impression of Change.
Patient-reported outcomes, as measured by PGIC and mCDQ-24, showed improvements over long-term treatment. The proportion of patients ‘Much Improved’ or ‘Very Much Improved’ on the PGIC was 54% (177/328) at Week 6, and 68% (44/65) at Week 80 (figure 4). The mean (SE) change in mCDQ-24 total score showed improvement in all patients from baseline to Weeks 15 (–5.0), 28 (–4.4) and 41 (–6.1). The mCDQ-24 overall mean (SE) change at Week 54 was –5.5 (1.2) (online supplementary table 3). Improvements in each of the five mCDQ-24 domains (ADL, emotional, pain, social and stigma) were also observed at Week 54 (online supplementary table 3).
Discussion
In this open-label extension of the ARM-TD15 and AIM-TD16 studies, treatment with deutetrabenazine at dosages from 12 mg/day to 48 mg/day was well tolerated and efficacious in patients with TD. These results confirm the safety and efficacy of deutetrabenazine demonstrated in the randomised, double-blind, placebo-controlled trials, and further show that these outcomes are maintained during long-term therapy.
Incidence rates of AEs during open-label treatment overall were comparable to or lower than those seen during short-term treatment in the ARM-TD and AIM-TD phase 3 trials. In clinical trials with TD patients, it is important to screen for depression and suicidality, regardless of treatment. Long-term deutetrabenazine treatment did not result in increased risk of suicidality or depression based on AE reporting or the C-SSRS assessment. There was a small increase in HADS depression subscale score at the end of treatment, although the clinical relevance is uncertain. Long-term extension data from prior studies on the earlier-generation compound tetrabenazine revealed a significant risk of drug-induced parkinsonism, although these studies were typically carried out in older patients compared with the patient population enrolled in this study.18–21 Overall, safety was maintained up to Week 106, suggesting that long-term exposure of deutetrabenazine did not result in cumulative toxicity or issues with tolerability; however, tolerability thresholds may differ in various patient populations, such as the elderly. These results further support the safety profile of deutetrabenazine, including use in patients with concomitant antipsychotic treatment, which may provide patients with a long-term treatment option that is not likely to exacerbate or interrupt the management of their underlying psychiatric conditions.
Long-term deutetrabenazine treatment also provided meaningful improvements in TD symptoms. While improvements in TD were noted by Week 2 based on the AIMS score, continued gradual improvement was seen through Week 106, which may be partially due to patient dropout. Similarly, a prior open-label study (mean follow-up time; 20.3 weeks, N=20) showed a 54.2% improvement in the mean AIMS score with tetrabenazine treatment.20 21 The improvement of TD symptoms observed in the present study was recognised by clinicians, who rated 45% of patients at Week 4 and 70% of patients at Week 80 as ‘Much Improved’ or ‘Very Much Improved’ according to the CGIC assessment. CGIC results for a previous open-label tetrabenazine study (mean follow-up time: 22 months) were consistent with these results, with 41.2% of patients reporting similar improvement, although the study was small (patients with TD: n=17).18 21 Improvement was not limited to that observed by clinicians; the majority of patients in our study also recognised improvement in their TD severity, as measured by the proportion of patients who reported themselves as ‘Much Improved‘ or ‘Very Much Improved‘ on the PGIC scale as early as Week 4 (44%), gradually increasing to 68% at Week 106. Quality of life measures, based on the mCDQ-24, showed improvements with long-term maintenance therapy. The consistent improvements across all efficacy assessments (AIMS, CGIC, PGIC and mCDQ-24) support the long-term clinical utility of deutetrabenazine for the treatment of TD.
This study’s open-label treatment design poses some inherent limitations. Given the lack of a control group, all outcomes were considered descriptive rather than inferential. This study used site-rated AIMS scores rather than the blinded central video ratings employed in the ARM-TD and AIM-TD studies. We cannot know whether the increased benefit over time was due to rating bias or whether it would still be apparent if blinded central video ratings or a control group had been used.
Overall, this open-label extension study demonstrated that deutetrabenazine was generally efficacious, safe and well tolerated in patients with TD, with improvements in AIMS scores, global improvement of TD severity, positive patient-reported outcomes and low rates of mainly mild-to-moderate AEs.
Acknowledgments
We thank the patients and site personnel involved with this study, and Kristen Hokenson, PhD (Chameleon Communications International with funding from Teva Pharmaceuticals) for editorial assistance in the preparation of this report.
References
Footnotes
Contributors HHF, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. DS, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. MDD, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. SAF, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. RAH, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. JJ-S, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. WGO, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. LFJ, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. SWW, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. DB, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. MSL, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. DRS, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript. KEA, significant content-related direction, data acquisition and analysis, contribution to the writing of the draft and feedback on all relevant materials throughout the development of the manuscript.
Funding This study was funded by Teva Pharmaceuticals, Petach Tikva, Israel.
Competing interests HHF has received honoraria from Prime Education, Inc, International Parkinson and Movement Disorders Society, Carling Communications, Medscape (speaker in CME events), AbbVie, Biogen, GE Health Care, Inventiv, Kyowa Hakko Kirin, Lundbeck, Merz Pharmaceuticals, Voyager, Sunovion, Pfizer Pharmaceuticals (as a consultant). He has received grant and research support from: AbbVie, Acadia, Teva, Biotie/Acorda Therapeutics, Civitas, Kyowa/Prostrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia; he has no owner interest in any pharmaceutical company. He has received royalties from: Demos Publishing (serving as a book author/editor). The Cleveland Clinic has a contract with Teva for his role as a Co-Principal Investigator in SD-809 tardive dyskinesia global studies. He also serves as a member of the publication committee for Acorda Pharmaceuticals but does not receive any personal compensation for this. He receives a stipend from the International Parkinson and Movement Disorders Society for serving as Medical Editor of the MDS Web Site. DS is a former employee of Teva Pharmaceuticals; he has received salary and benefits from Auspex Pharmaceuticals, and has patents pending: US20160346270, US20160287574. MDD is an employee of Teva Pharmaceuticals. SAF has received honoraria from Neurocrine, Lundbeck, Teva, Avanir, UCB, US WorldMeds, Sunovion, Adamas; he has received research support from Ipsen, Medtronic, Auspex, US WorldMeds, Pharm-Olam, Cynapsus Therapeutics/Sunovion, Vaccinex, Solstice, CHDI Foundation, Michael J. Fox Foundation, NIH; he has received royalties from Demos, Blackwell Futura for textbooks, and UpToDate. RAH has served as a consultant for Teva Pharmaceuticals, AbbVie, Inc, Acorda Therapeutics, Adamas Pharmaceuticals, AstraZeneca, Biotie Therapies, Cynapsus Therapeutics, Impax Laboratories, Inc, Lundbeck LLC, Michael J Fox Foundation, Neurocrine Biosciences, Neuropore Therapies, Pfizer Inc, Prexton Therapeutics, US WorldMeds, Guidepoint Global, Gerson Lehrman Group (GLG), LCN Consulting, Putnam Associates, National Parkinson Foundation, eResearch Technology, Inc, Sarepta Therapeutics, Back Bay Life Science, National Institutes of Health, Projects in Knowledge, Vista Research, LifeMax, Peerview Press, ClinicalMind Medical and Therapeutic Communications, Sunovion Pharmaceuticals, Academy for Continued Healthcare Learning, Outcomes Insights, Expert Connect, HealthLogix, Cowen and Company, Pharma Two B, Ltd, RMEI Medical Education for Better Outcomes, ClearView Healthcare Partners, Health Advances, Kyowa Kirin Pharmaceutical Development, Ltd, Quintiles, and Eli Lilly and Company. JJ-S has received research support from Acadia Pharmaceuticals, St. Jude Medical, Biotie, Michael J. Fox Foundation; she has served as a consultant for Teva, St. Jude Medical, Medtronic; she has also received honoraria from Vindico Medical Education. WGO has received research support from Lundbeck, Tremor Research Group, Dystonia Coalition, Restless Leg Syndrome Foundation, Acorda. Speakers’ Bureau: Teva, Lundbeck, UCB, US WorldMeds; he has served as a consultant for Teva, Lundbeck, Acadia. LFJ has received grant support Auspex/Teva, Boehringer Ingelheim, Otsuka, NIH; he has served as a consultant for Roche. SWW has received research support from Teva, Pfizer; he has served as a consultant for Boehringer Ingelheim, Nutria Health. DB has received research support from National Parkinson Foundation; he has served on the speaker’s bureau for Teva Pharmaceuticals and Acadia Pharmaceuticals, Inc; he has served as a consultant for Teva, Cynapsus, Acadia. MSL has served on the speakers’ bureau for Lundbeck, Acadia, and Teva; he has served as a consultant for Teva Neuroscience, US WorldMeds, the Mayo Clinic, Starnes-Davis-Florie, Elite Medical Experts, Spears, Moore, Rebman receives research support from the National Institutes of Health (R01 NS082296, R21 GM118962, U54 TR001456, 5U01 NS090259), Department of Defense (W81XWH-17-1-0062), Michael J. Fox Foundation, Dorothy/Daniel Gerwin Parkinson’s Research Fund, Auspex, Teva, Acorda, US WorldMeds, Axovant and CHDI; and receives royalty payments from Elsevier for editing ‘Animal Models of Movement Disorders’ and ‘Movement Disorders: Genetics and Models.’ DRS has served as a consultant for Eli Lilly, Teva, Lundbeck, and Weston Brain Institute; he has received speaker’s fees from Acadia, Baylor College of Medicine, the Arizona Psychiatric Society, Lundbeck, Teva US World Meds and the Tourette Association of America; he has received research support from Arizona Alzheimer’s Consortium, Axovant, Biotie, Intec, Kyowa, Teva, US WorldMeds, Neurocrine, Michael J. Fox Foundation, and the NIH. KEA has served as a scientific adviser and was the North American study Co-Principal Investigator for LEGATO-HD, Global Principal Investigator for AIM-TD, and Global Co-Principal Investigator for ARM-TD; she was the site Principal Investigator for Pride-HD, First-HD, ARC-HD for Teva; she was the scientific advisor and site Principal Investigator for ENROLL-HD for CHDI Foundation; she was a scientific advisor for Prana and site Principal Investigator for Vaccinex; she served as a consultant for the NeuroNext 105 study for Azevan; she has received salary support from the Griffin Foundation. She has also received honoraria from Vindico Medical Education.
Patient consent for publication Obtained.
Ethics approval Before this study was initiated, the protocol was submitted to the Independent Ethics Committee (IEC)/Institutional Review Board (IRB) according to national or local regulations. Any protocol amendments were also submitted. Written informed consent was obtained from each patient before any study procedures or assessments were done.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.