Article Text

Abstract
Objective To investigate the efficacy of rovatirelin, a thyrotropin-releasing hormone analogue, for ataxias in patients with spinocerebellar degeneration (SCD).
Methods Two multicentre, randomised, double-blind, placebo-controlled phase 3 studies (KPS1301, KPS1305) enrolled patients with predominant cerebellar ataxia, including SCA6, SCA31 or cortical cerebellar atrophy. KPS1301 enrolled patients with truncal ataxia and KPS1305 enrolled patients with truncal and limb ataxia. Each study included 4 weeks of pretreatment, a 28-week or 24-week treatment period and 4 weeks of follow-up. Patients were randomised (1:1:1) to rovatirelin (1.6 or 2.4 mg) or placebo in KPS1301, and randomised (1:1) to rovatirelin 2.4 mg or placebo in KPS1305. The primary endpoint was change in Scale for the Assessment and Rating of Ataxia (SARA) total scores. Pooled analysis was performed in patients who met the SARA recruitment criteria of KPS1305.
Results From October 2013 to May 2014, KPS1301 enrolled 411 patients; 374 were randomised to rovatirelin 1.6 mg (n=125), rovatirelin 2.4 mg (n=126) or placebo (n=123). From November 2016 to August 2017, KPS1305 enrolled 241 patients; 203 were randomised to rovatirelin 2.4 mg (n=101) or placebo (n=102). The primary endpoint showed no significant difference between rovatirelin and placebo in these two studies. In the pooled analysis (n=278), the difference between rovatirelin 2.4 mg (n=140) and placebo (n=138) was –0.61 (–1.64 vs –1.03; 95% CI –1.16 to –0.06; p=0.029) in the adjusted mean change in the SARA total score.
Conclusions Rovatirelin is a potentially effective treatment option for SCD.
Trial registration number NCT01970098; NCT02889302
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Spinocerebellar degeneration (SCD) is a neurodegenerative disease that is characterised by progressive cerebellar ataxia.1 2 The main symptoms of SCD are gait ataxia, standing instability, limb ataxia and dysarthria.3 In addition, pyramidal tract, extrapyramidal or peripheral nervous symptoms can occur in patients with some types of SCDs.3 Hereditary SCA6, SCA31 and sporadic cortical cerebellar atrophy (CCA) mainly show cerebellar symptoms, while hereditary SCA1, SCA2, SCA3 (Machado-Joseph disease) and sporadic multiple system atrophy (MSA) show cerebellar symptoms and extracerebellar symptoms. The presence of extracerebellar symptoms also contributes to a patient’s disability and may interfere with evaluation of the severity of cerebellar ataxia. Therefore, it is considered more useful to evaluate the effect of a drug on cerebellar ataxia in patients with predominant cerebellar symptoms.
Thyrotropin-releasing hormone (TRH) is a hypothalamic hormone that promotes thyroid-stimulating hormone and prolactin (PRL) release from the pituitary gland, and acts broadly on the central nervous system to activate several neurotransmitters.4–7 TRH has been shown to improve ataxia in an ataxia mouse model with a CACNA1A mutation, the causative gene for SCA6.8 9 In a 2-week, double-blind, placebo-controlled trial in 254 SCD patients, the efficacy of a TRH analogue (TRH tartrate) was demonstrated in patients with predominantly ataxic forms of SCD.10 Thus, the first TRH analogue was approved in Japan in 1985 for the treatment of ataxia associated with SCD. However, in the previous clinical trial, the diagnostic criteria for SCD were not consistent with recent diagnostic criteria. Further, over 60% of patients with the predominantly ataxic forms of SCD had olivopontocerebellar atrophy, and the drug effects were not assessed using an ataxia rating scale alone. Finally, the duration of the trial was only 2 weeks. Therefore, well-designed double-blind trials were considered necessary to demonstrate the efficacy of new drug candidates for cerebellar ataxia.11 12
Rovatirelin is a new TRH analogue,13 which showed higher affinity for human TRH receptors and greater absorption and transition into and stability in the brain than the existing TRH analogue, taltirelin.14 15 The effects of rovatirelin on the ataxic rolling Nagoya mice, which carry a mutation in the CACNA1A gene, were more potent and lasted longer than those of taltirelin (data on file). To investigate the efficacy and safety of rovatirelin for the treatment of cerebellar ataxia, we performed two large-scale, randomised, double-blind, phase 3 studies in patients with predominant cerebellar symptoms of SCD.
Methods
Study design and participants
Two similar multicentre, randomised, double-blind, placebo-controlled, phase 3 studies (KPS1301 and KPS1305) were conducted in Japanese patients with SCD. Patients were enrolled from 86 hospitals in Japan between 9 October 2013 and 30 May 2014 in KPS1301 and from 62 hospitals in Japan between 15 November 2016 and 1 August 2017 in KPS1305.
Both studies included patients aged ≥20 years with only predominant cerebellar symptoms of SCD (CCA or genetically confirmed SCA6 or SCA31). The diagnosis of CCA was mainly based on the idiopathic cerebellar ataxia diagnostic criteria proposed by Yoshida et al.16 Briefly, patients with slowly progressive adult-onset cerebellar ataxia and with no first-degree or second-degree relatives with ataxia were enrolled, while patients with autoimmune-mediated, metabolic, alcoholic and drug-induced ataxia were excluded. In addition, patients with MSA were carefully excluded based on the second consensus statement for the diagnosis of MSA.17 A detailed diagnostic flow diagram of CCA is shown in online supplementary figure 1.
Supplemental material
The KPS1301 study included patients with truncal ataxia (Scale for the Assessment and Rating of Ataxia (SARA)18 19 total score of ≥6 and SARA gait score of 2–6). The KPS1305 study included patients with both truncal and limb ataxia (SARA gait score of 2–6, stance score of ≥2 and scores of ≥1 for the finger chase, nose-finger and fast alternating hand movements tests (patients with scores of ≥1 on unilateral tests were included if tests were not feasible on the right or left side for some reason other than the primary disease)).
Exclusion criteria were: secondary ataxia (eg, cerebrovascular disorder, brain tumour, multiple sclerosis, hypothyroidism, drug-induced, paraneoplastic syndrome); suspected alcoholic ataxia; motor disorder due to musculoarticular disease; cognitive impairment, depression or other mental diseases and a diagnosis of thyrotoxicosis. In the KPS1305 study, patients who had participated in a rovatirelin study ≥2 years ago were eligible to enrol; however, this group of patients was restricted to ≤30% of the total number of patients enrolled in KPS1305.
Randomisation and masking
Patients were randomised using an Interactive Web Response System by an independent organisation. They received rovatirelin 1.6 mg, rovatirelin 2.4 mg or placebo (1:1:1) in the KPS1301 study, and rovatirelin 2.4 mg or placebo (1:1) in the KPS1305 study. In KPS1305, the presence/absence of participation in a previous clinical study of rovatirelin was included as a stratification factor for dynamic allocation. All patients and personnel involved in the study, including the investigators and study-site staff, were masked to the treatment assignment. The test agents were indistinguishable from one another, and indistinguishability was confirmed at the time of test agent allocation and at the end of the study. Results of the endocrinology tests were stored at the measuring institution until unblinding.
Procedures
Both studies included a single-blind 4-week pretreatment period, during which only the patients were blinded and were given oral placebo once daily after breakfast. Thereafter, under double-blind conditions, eligible patients received oral rovatirelin or placebo once daily (after breakfast) for 28 weeks (KPS1301) or 24 weeks (KPS1305). In a follow-up period of 4 weeks, the patients were given no test agents and underwent only safety evaluation. TRH analogues, CYP3A4 inhibitors and P-glycoprotein inhibitors were prohibited during the pretreatment and treatment periods. In addition, investigational products other than rovatirelin were prohibited throughout the study.
Efficacy and safety were evaluated during hospital visits every 4 weeks. At weeks 0 through 24, SARA (Japanese version) was used to assess the degree of ataxia. Each patient was assessed for SARA by the same investigator, who was a board-certified neurologist of the Japanese Society of Neurology. Because differences in SARA assessment between investigators cannot be eliminated in large-scale, multicentre trials, the investigators were trained using a video of the SARA assessment method to reduce the variability in SARA assessments. Patient quality of life (QoL) was self-assessed using the Japanese version of the Short Form-8 (SF-8) questionnaire at weeks 0 through 24.20
Outcomes
The primary endpoint was change in the SARA total score from baseline to the time point of final evaluation. The final evaluation was defined as the latest evaluation, excluding missing data. Secondary efficacy endpoints were the SARA total score, individual SARA scores and SF-8 scores at each evaluation time point. Safety endpoints included adverse events (AEs), vital signs, body weight, 12-lead ECG, clinical laboratory tests and endocrinology tests (thyroid stimulating hormone (TSH), free triiodothyronine (FT3), free thyroxine (FT4) and PRL). Investigators evaluated AEs for severity (mild, moderate or severe) and for causal relationships with the test agents.
Statistical analyses
For the KPS1301 study, assuming a difference in the change in the SARA total score between the rovatirelin group and the placebo group of −0.75, and a common SD of 2.0, a sample size of 113 randomly assigned patients was calculated as sufficient to provide 80% power with a two-sided 5% significance level. In the KPS1305 study, a sample size of 87 randomly assigned patients was calculated as sufficient to provide 80% power with a two-sided 5% significance level, assuming a difference in the change in the SARA total score between the rovatirelin 2.4 mg group and the placebo group of −1.06 and a common SD of 2.48.
The full analysis set, which was used for efficacy analysis, excluded patients who were untreated or ineligible, violated the Good Clinical Practice (GCP), discontinued before the beginning of the treatment period or were unavailable for the primary endpoint. The safety set, which was used for safety analysis, excluded patients who violated the GCP, were untreated or discontinued before the beginning of the treatment period.
The primary endpoint in both studies and the pooled analysis was calculated as the difference in SARA total score between the final evaluation and week 0 (the end of the pretreatment period). The change in SARA total score between the two groups was compared using analysis of covariance, with the treatment group as the fixed effect and the SARA total score at week 0 as the covariate in KPS1301. The presence/absence of participation in a previous clinical study of rovatirelin was also included in KPS1305. In KPS1301, a closed testing procedure was used for adjustment of multiplicity, and superiority of rovatirelin 1.6 mg over placebo was verified only after superiority of rovatirelin 2.4 mg over placebo was confirmed.
We hypothesised that patients with less severe ataxia show a more prominent placebo effect. Therefore, a subgroup analysis of the primary endpoint was performed in patients in the KPS1301 study who met the KPS1305 inclusion criteria (SARA gait score of 2–6, stance score of ≥2 and scores of ≥1 for the finger chase, nose–finger and fast alternating hand movements tests).
A pooled retrospective analysis was performed that combined the data from the above subgroup of KPS1301 and from all patients in KPS1305 with the same inclusion criteria (SARA gait score of 2–6, stance score of ≥2 and scores of ≥1 for the finger chase, nose–finger and fast alternating hand movements tests). The patients who received either rovatirelin 2.4 mg or placebo and had not participated in a previous rovatirelin study were included. Furthermore, in the pooled analysis, two subgroup analyses were also performed in patients with a baseline SARA total score of ≥15 and <15, because the median baseline SARA total score in the pooled group was 15.
The significance level for each two-sided statistical testing was 5%. In the comparison of efficacy variables between the two groups, two-sample t-tests were used for continuous variables. All analyses used SAS V.9.4 (SAS Institute).
Results
Patient population
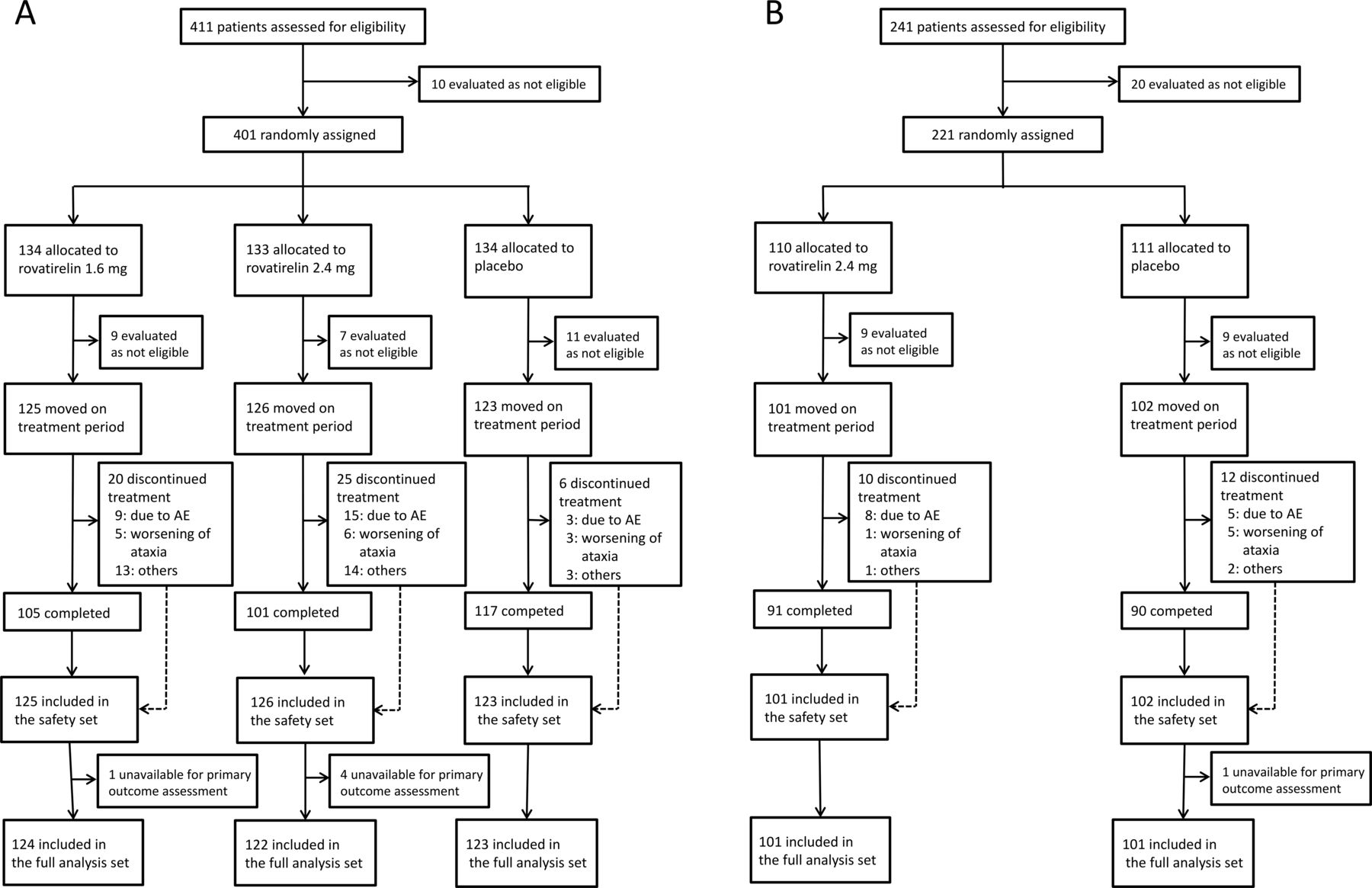
In the KPS1301 study, 411 patients with truncal ataxia were enrolled; 374 of these patients were included in the safety set and 369 in the full analysis set (figure 1A). In the KPS1305 study, 241 patients with truncal and limb ataxia were enrolled; 203 were included in the safety set and 202 in the full analysis set (figure 1B). The pooled analysis included 278 patients; 140 in the rovatirelin 2.4 mg group and 138 in the placebo group. In both studies, there were no major between-group differences in baseline characteristics (table 1). In KPS1301, 165 patients had SCA6, 72 patients had SCA31 and 132 patients had CCA; respective numbers in KPS1305 were 83 (SCA6), 57 (SCA31) and 62 (CCA) patients. The mean baseline SARA total score was 12.9 in KPS1301 and 14.9 in KPS1305. In addition, the mean baseline SARA total score was lower for patients with truncal and limb ataxia in the KPS1305 study versus patients with truncal and limb ataxia in the KPS1301 study (14.9 vs 16.8, p<0.001, two-sample t-test). The SARA total scores during the pretreatment period were stable for patients who received treatment in the two trials (n=577; change in SARA total score during the pretreatment period: 0.04±1.59, mean±SD, p=0.530, one-sample t-test).
Baseline demographics and characteristics of the full analysis set

Flow chart of patients included in the (A) KPS1301 and (B) KPS1305 studies. The reasons for discontinuation could overlap. AE, adverse event.
Treatment was discontinued in KPS1301 by 16.0% of patients receiving rovatirelin 1.6 mg, 19.8% of patients receiving 2.4 mg and 4.9% of patients receiving placebo (figure 1A). In KPS1305, treatment was discontinued by 9.9% of patients in the rovatirelin 2.4 mg group and 11.8% of patients in the placebo group (figure 1B). The main reasons for the discontinuation were AEs and worsening of ataxia (figure 1).
Efficacy
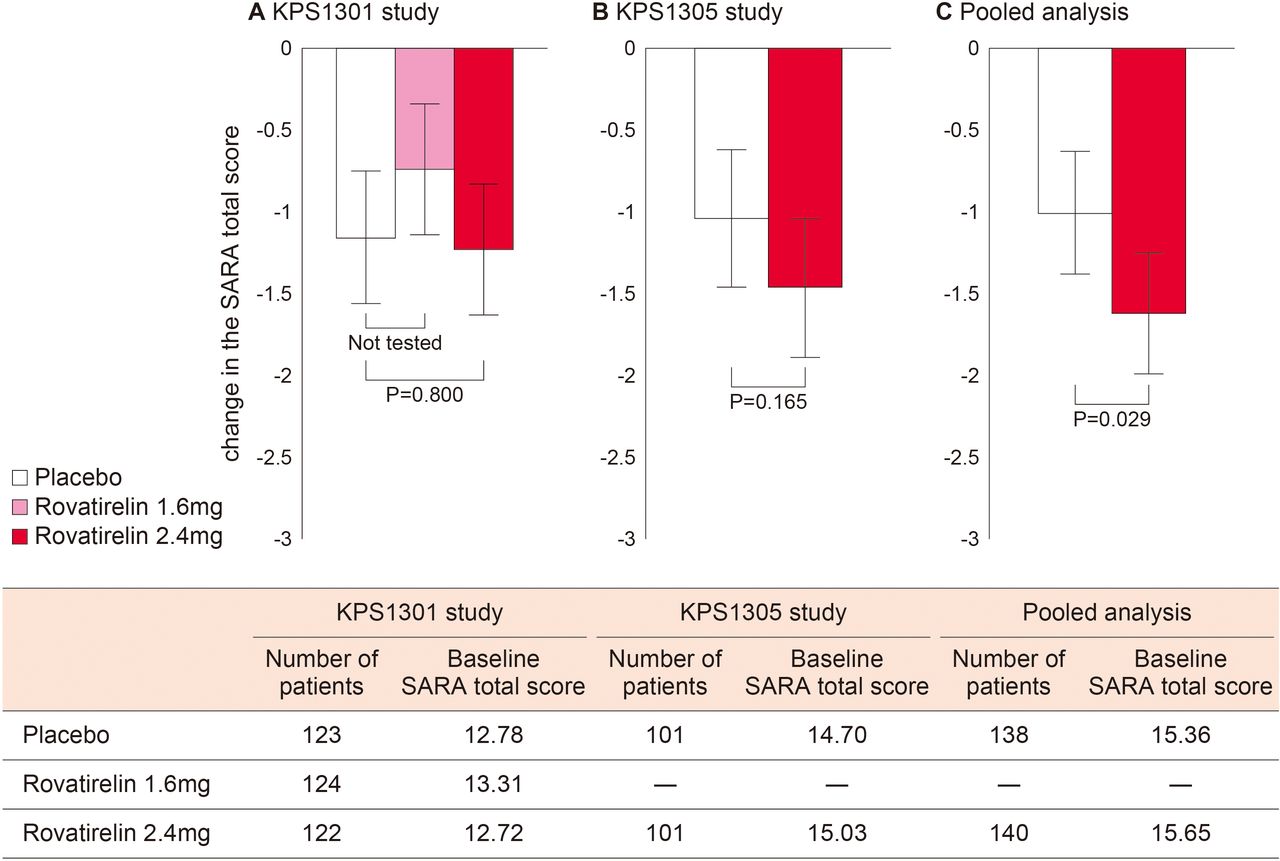
In KPS1301, the adjusted mean change in SARA total score at the final evaluation time point (primary endpoint) was −0.74 for rovatirelin 1.6 mg, −1.23 for rovatirelin 2.4 mg and −1.16 for placebo; the difference between rovatirelin 2.4 mg and placebo (−0.07; 95% CI −0.64 to 0.49) was not significant (p=0.800, figure 2A).

Adjusted mean and 95% CI of the change in SARA total score from baseline (primary endpoint) in the (A) KPS1301 and (B) KPS1305 studies. P values were calculated using the ANCOVA method. (C) Pooled analysis of patients from KPS1301 and KPS1305 studies who met the KPS1305 inclusion criteria (SARA gait score of 2–6; stance score of ≥2; finger chase, nose–finger and fast alternating hand movement scores of ≥1 each). ANCOVA, analysis of covariance; SARA, Scale for the Assessment and Rating of Ataxia.
In the subgroup of patients with limb and truncal ataxia (n=136, online supplementary table 1), the adjusted mean change in SARA total score at the final evaluation time point was −0.65 for rovatirelin 1.6 mg, −1.98 for rovatirelin 2.4 mg and −0.92 for placebo. In this subgroup, the difference between rovatirelin 2.4 mg and placebo was significant (−1.06; 95% CI −2.08 to –0.04; p=0.041).
Based on these results, an additional trial in patients with limb and truncal ataxia was conducted (KPS1305). In KPS1305, the adjusted mean change in SARA total score showed no significant difference between rovatirelin 2.4 mg and placebo (−1.46 vs −1.04; difference −0.42; 95% CI −1.02 to 0.18; p=0.165; figure 2B).
To obtain a more precise estimate of rovatirelin treatment effects and greater statistical power for assessment,21 data from KPS1305 and the KPS1301 subgroup were combined, because the study protocols for both groups were identical except for the SARA score criteria. The baseline characteristics of the pooled group (n=278) are shown in table 1. In this pooled analysis, there was a significant difference in the adjusted mean change in SARA total score between rovatirelin 2.4 mg and placebo (−1.64 vs −1.03; difference −0.61; 95% CI −1.16 to –0.06; p=0.029; figure 2C). In individual SARA scores, significantly greater reductions in fast alternating hand movement scores were observed with rovatirelin 2.4 mg vs placebo (−0.34 vs −0.08; p<0.001).
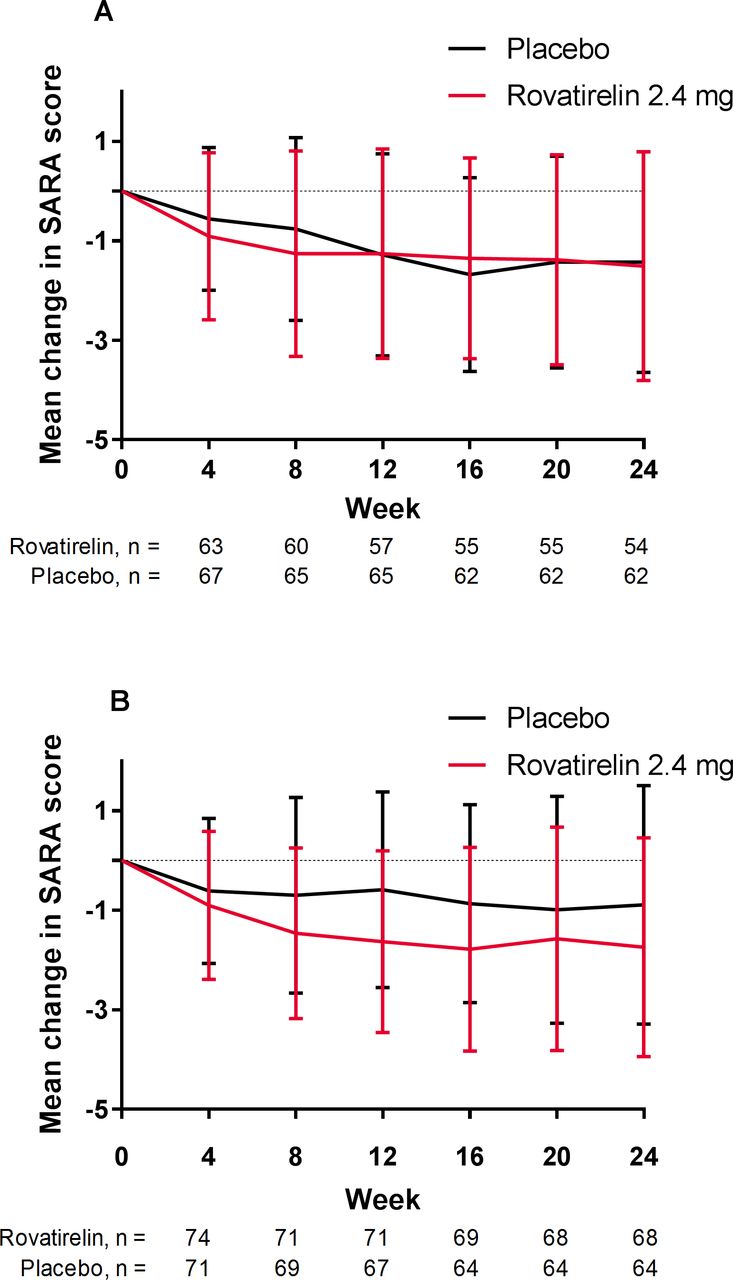
To test whether rovatirelin was effective in patients with more severe ataxia, the pooled analysis group was divided into two groups using the median baseline SARA total score of 15 as the severity threshold. Patients with a baseline SARA total score ≥15 showed a significantly greater adjusted mean change in SARA total score with rovatirelin 2.4 mg compared with placebo (−1.75 vs −0.58; difference −1.16; 95% CI −1.95 to –0.38; p=0.003). In patients with baseline SARA total score <15, the difference in adjusted mean change in SARA total score between rovatirelin 2.4 mg and placebo was not significant (−1.54 vs −1.48; difference −0.06; 95% CI −0.85 to 0.73; p=0.879). In the placebo group, greater score reductions were observed in patients with a baseline SARA total score <15 than in those with a baseline score ≥15, whereas scores were similar between patients with SARA total score <15 and ≥15 in the rovatirelin 2.4 mg group (figure 3). In patients with SARA total score ≥15, individual SARA scores were significantly reduced with rovatirelin 2.4 mg versus placebo for gait (−0.18 vs 0.13; p=0.025) and fast alternating hand movement (−0.49 vs −0.11, p=0.001).
{kind=link}
{kind=link}
{kind=link}
Mean±SD change in SARA total score from baseline for patients in the pooled populations with a baseline SARA score of (A) <15 or (B) ≥15. SARA, Scale for the Assessment and Rating of Ataxia.
There were no marked changes from baseline in the SF-8 physical health summary score or mental health summary score in the QoL evaluation for any of the treatment groups (table 2).
Secondary endpoints
Safety
The most common treatment-emergent AEs (occurring in ≥5% of patients in both studies) with rovatirelin were nasopharyngitis, nausea, weight loss and contusion (table 3). Common AEs considered to be related to rovatirelin (occurring in ≥5% of patients in any rovatirelin group) were weight loss, nausea and decreased appetite. The majority of AEs were judged to be mild in severity (table 3). AEs leading to treatment discontinuation in 7.2%, 11.9% and 2.4% of patients receiving rovatirelin 1.6 mg, rovatirelin 2.4 mg and placebo, respectively, in KPS1301 (p=0.011, Fisher’s exact test) and 7.9% and 4.9% of patients receiving rovatirelin 2.4 mg and placebo, respectively, in KPS1305 (p=0.407, Fisher’s exact test; table 3).
Adverse events (AEs)
In patients receiving rovatirelin, mean TSH, FT3, FT4 and PRL levels increased from baseline to week 4; however, the levels were not further increased with continued administration of rovatirelin. These hormone levels exceeded the normal range at some evaluation time points, and mean FT4 levels exceeded the reference value at all evaluation time points (online supplementary tables 2 and 3). Mean FT4 levels returned to baseline values during the follow-up period. The body weight of patients decreased from baseline to final evaluation in each of the rovatirelin groups in KPS1301 (–2.00 kg in the 1.6 mg group, –2.16 kg in the 2.4 mg group and –0.28 kg in the placebo group; p<0.001, one-way analysis of variance) and in the rovatirelin group in KPS1305 (–2.31 kg in the 2.4 mg group vs 0.13 kg in the placebo group; p<0.001 two-sample t-test). No clinically significant changes or findings were observed in vital signs, 12-lead ECG or clinical laboratory tests.
Discussion
We found no significant differences in efficacy between rovatirelin and placebo in two prospective randomised controlled studies in patients with predominant cerebellar symptoms of SCD. However, in the pooled analysis of data from the two studies, rovatirelin 2.4 mg was associated with a significantly greater reduction in SARA total score compared with placebo (−1.64 vs −1.03; p=0.029) in patients with predominant cerebellar symptoms of SCD. This improvement was more prominent in patients with relatively severe ataxia versus less severe ataxia (−1.75 vs −0.58; p=0.003). We consider this pooled analysis to be valid for the following reasons: (1) pooled data were obtained from identically designed studies; (2) the two studies were conducted within a relatively short period and (3) there was no heterogeneity of patient background between studies (online supplementary figure 2). This pooled analysis allowed us to obtain a more precise estimate of rovatirelin treatment effects and greater statistical power for assessment,21 indicating that rovatirelin may be a potentially effective treatment option for cerebellar ataxia in patients with predominant cerebellar symptoms of SCD.
The effect of rovatirelin observed in our two individual studies was not significant and showed limited clinical relevance during the 24 weeks treatment period. In fact, both phase 3 studies did not show significant improvements in SF-8 and QoL. However, progression of SCD is generally slow. It has been reported that the annual changes in SARA total score are 0.80–1.33 for SCA6 and 0.8 for SCA31.2 22 23 Considering the sensitivity of SARA and slow progression, limited change in SARA total score was expected during the observation period selected for our study. Furthermore, it should be noted that QoL assessments require long-term observation periods.
Although the patient inclusion criteria for the KPS1305 study were revised based on the positive outcomes of the KPS1301 subgroup analysis, the KPS1305 study did not show any treatment difference between rovatirelin and placebo. Patients in KPS1305 had lower baseline SARA total scores than those in the KPS1301 subgroup (14.9 and 16.8, respectively, p<0.001). Therefore, we hypothesised that patients with less severe ataxia show a more prominent placebo effect. In fact, among patients in the placebo groups, greater SARA score reductions were observed in patients with less severe ataxia than in those with severe ataxia (−1.48 vs −0.58, respectively). On the other hand, in patients receiving rovatirelin, a similar decrease in SARA total score was observed in both groups (−1.54 vs −1.75, respectively). These findings suggest that rovatirelin efficacy was not observed in the KPS1305 study due to the inclusion of a substantial number of patients with mild ataxia and a prominent placebo effect.
It is well known that clinical trials for neurodegenerative diseases often observe a large placebo effect. Our study indicates that the placebo effect should be taken into consideration when evaluating the efficacy of treatments for cerebellar ataxia. Our findings also suggest that the placebo effect can be decreased by restricting treatment to patients with relatively severe ataxia when developing clinical trial protocols for cerebellar ataxia. Although the reason for the prominent placebo effect observed in patients with mild ataxia is unknown, patients with baseline SARA total scores <15 were generally younger and had a shorter disease duration compared with patients with baseline SARA total scores ≥15. One possible reason for this could be that patients with a shorter disease duration may have expected better improvement with treatment received in the trial. These factors should be kept in mind and carefully considered in clinical trials of ataxia in the future.
In our studies, rovatirelin was associated with increases in thyroid hormone levels to near the upper limits of the reference ranges. Most AEs (eg, nausea and weight loss) were considered to be attributable to these increases in thyroid hormones. However, severe or serious AEs were rare. Treatment discontinuation was more frequent with rovatirelin treatment versus placebo. Common AEs leading to treatment discontinuation were nausea, decreased appetite and insomnia, all of which were of mild or moderate severity. Thus, rovatirelin was considered to have an acceptable tolerability profile. However, long-term AEs with chronic rovatirelin use need to be monitored in future studies.
Drugs for neurodegenerative disease are expected to halt degeneration and improve symptoms.1 Mechanistically, as a TRH analogue, rovatirelin can improve motor function by increasing various neurotransmitter levels9 and having a neuroprotective effect.24 In fact, in a pharmacological study in rats, rovatirelin increased locomotor activity associated with increased noradrenaline concentration,14 as a result of its rapid absorption, stability in plasma and transition into the brain.15 Further investigation is necessary to examine whether rovatirelin can inhibit the progression of SCD with long-term treatment.
Our studies only included patients with predominant cerebellar symptoms of SCD. The molecular pathogenesis of cerebellar degeneration in predominant cerebellar symptoms of SCD includes abnormal elongation of CAG repeat sequences in the CACNA1A gene in SCA6,25 augmentation of the (TGGAA)n sequence located in an intron in SCA3126 and mixed mechanisms in CCA.27 The fact that rovatirelin showed efficacy in these conditions indicates that this drug exerts a beneficial effect on cerebellar function as a whole. This suggests that rovatirelin may improve motor function in patients with extracerebellar symptoms of SCD.
Our studies have several limitations. Although improvements in SARA total score were found in pooled and subgroup analyses, these analyses were post hoc in nature. The studies included only Japanese patients, which may influence the generalisability of results. Although the number of repeats can influence the clinical severity of SCA6 and SCA31, data on the number of repeats were not obtained. However, as the effect of the number of repeats on the pathological condition is often subtle in these diseases, this lack of data was considered to be inconsequential.2 23 28 As genetic analyses were not essentially required for CCA diagnosis, it is possible that patients with SCA6, SCA31 or other genetic disorders were included in these studies.
Conclusion
In two large-scale double-blind studies in patients with predominant cerebellar symptoms of SCD, rovatirelin was not associated with a statistically significant change in SARA total score compared with placebo; however, the pooled analysis of patients with both truncal and limb ataxia indicated that rovatirelin is a potentially effective treatment option against cerebellar ataxia in patients with predominant cerebellar symptoms of SCD. This improvement was more prominent in patients with relatively severe ataxia than in those with less severe ataxia. Further studies are needed to assess the long-term efficacy and safety of rovatirelin, as well as patient-reported outcomes.
Acknowledgments
Statistical analyses were completed by MY, Kissei Pharmaceutical, Bunkyo-ku, Tokyo, Japan. We thank Yoshiko Okamoto, inScience Communications, Springer Healthcare for providing editorial support and Toni Dando and Sarah Greig, of inScience Communications, Springer Healthcare for editing and styling the manuscript for submission. This medical writing assistance was funded by Kissei Pharmaceutical.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it appeared Online First. 'Department of Neurology' was removed from the corresponding author address. In the sentence "In the subgroup of patients with limb and truncal ataxia…", 'n=13690' has been corrected to 'n=136'.
Collaborators The Rovatirelin Study Group included following physicians: Takao Hashimoto (Aizawa Hospital), Masashiro Sugawara (Akita University Hospital), Masahiko Tomiyama (Aomori Prefectural Central Hospital), Kazuhiro Takamatsu (Brain Attack Center Ota Memorial Hospital), Masaru Kuriyama (Brain Attack Center Ota Memorial Hospital), Shigeki Hirano (Chiba University Hospital), Takahide Nagashima (Dokkyo Medical University Hospital), Kensho Okamoto (Ehime Prefectural Central Hospital), Ryuji Saigo (Fujimoto General Hospital), Ryuichi Okubo (Fujimoto General Hospital), Akira Iwasaki (Fukaya Red Cross Hospital), Hiroyuki Enomoto (Fukushima Medical University Hospital), Akio Kimura (Gifu University Hospital), Yoshio Ikeda (Gunma University Hospital), Mikio Shoji (Hirosaki University Hospital), Tetsuya Takahashi (Hiroshima University Hospital), Tsuneo Fujita (Hitachi General Hospital), Ichiro Yabe (Hokkaido University Hospital), Hidenao Sasaki (Hokkaido University Hospital), Asako Takei (Hokuyukai Neurological Hospital), Hirotaka Shimizu (Hyogo Brain and Heart Center), Hiroo Yoshikawa (Hyogo College of Medicine), Tatsui Nagadou (Imakiire General Hospital), Katsuhiko Kayanuma (Ina Central Hospital), Tomoko Ogawa (International University of Health and Welfare Hospital), Masanori Mizuno (Iwate Medical University Hospital), Yoji Goto (Japanese Red Cross Nagoya Daiichi Hospital), Hiroyuki Yahikozawa (Japanese Red Cross Society Nagano Hospital), Takuji Yasude (Japanese Red Cross Society Suwa Hospital), Akiyo Hineno (Japanese Red Cross Society Suwa Hospital), Kazuma Kaneko (Japanese Red Cross Society Suwa Hospital), Haruo Shimazaki (Jichi Medical University Hospital), Kazuyuki Noda (Juntendo University Shizuoka Hospital), Kazushi Deguchi (Kagawa University Hospital), Hiroshi Takashima (Kagoshima University Hospital), Tatsuhiko Yuasa (Kamagaya General Hospital), Makoto Matsui (Kanazawa Medical University Hospital), Satoshi Kaneko (Kansai Medical University Hirakata Hospital), Yoshihide Sunada (Kawasaki Medical School Hospital), Susumu Kusunoki (Kindai University Hospital), Yusaku Nakamura (Kindai University Sakai Hospital), Kazutoshi Nishiyama (Kitasato University Kitasato Institute Hospital), Tatsushi Toda (Kobe University Hospital), Kenji Sekiguchi (Kobe University Hospital), Yasushi Osaki (Kochi Medical School Hospital), Yasushi Maeda (Kumamoto Saishunso National Hospital), Mika Jikumaru (Kumamoto University Hospital), Satoshi Yamashita (Kumamoto University Hospital), Hirofumi Yamashita (Kyoto University Hospital), Takakuni Maki (Kyoto University Hospital), Taira Uehara (Kyushu University Hospital), Ryo Yamasaki (Kyushu University Hospital), Hiroo Yamaguchi (Kyushu University Hospital), Shinji Ohara (Matsumoto Medical Center), Akinori Nakamura (Matsumoto Medical Center), Akira Taniguchi (Mie University Hospital), Atsushi Inoue (Nagano Prefectural Kiso Hospital), Naoki Atsuta (Nagoya University Hospital), Masahisa Katsuno (Nagoya University Hospital), Yuji Takahashi (National Center of Neurology and Psychiatry), Sagiri Isose (National Hospital Organization Chiba-East-Hospital), Katsuhisa Ogata (National Hospital Organization East Saitama National Hospital), Naonobu Futamura (National Hospital Organization Hyogo-Chuo Hospital), Ryoichi Kurisaki (National Hospital Organization Kumamoto Minami Hospital), Taro Yamashita (National Hospital Organization Kumamoto Minami Hospital), Yoshito Sonoda (National Hospital Organization Minami Kyushu National Hospital), Ryusuke Matsumura (National Hospital Organization Nara Medical Center), Takashi Nakajima (National Hospital Organization Niigata National Hospital), Ryoko Koike (National Hospital Organization Nishi-Niigata Chuo Hospital), Tokio Shimomura (National Hospital Organization Saigata Medical Center), Hideyuki Sawada (National Hospital Organization Utano Hospital), Satoshi Kamei (Nihon University Itabashi Hospital), Shuichi Igarashi (Niigata City General Hospital), Kunihiko Makino (Niigata Prefectural Shibata Hospital), Masayoshi Tada (Niigata University Medical & Dental Hospital), Masayuki Ueda (Nippon Medical School Hospital), Mineo Yamazaki (Nippon Medical School Hospital), Hiroshi Nagayama (Nippon Medical School Hospital), Youichi Hokezu (Oita Prefectural Hospital), Etsuro Matsubara (Oita University Hospital), Yasuhiro Aso (Oita University Hospital), Koji Abe (Okayama University Hospital), Fumiharu Kimura (Osaka Medical College Hospital), Shigeki Arawaka (Osaka Medical College Hospital), Makio Takahashi (Osaka Red Cross Hospital), Kuni Konaka (Osaka University Hospital), Kousuke Baba (Osaka University Hospital), Kazuko Hasegawa (Sagamihara National Hospital), Jun Miki (Saku Central Hospital), Chinatsu Kobayashi (Saku Central Hospital), Yasutaka Tajima (Sapporo City General Hospital), Shin Hisahara (Sapporo Medical University Hospital), Shuichi Ikeda (Shinshu University Hospital), Yoshiki Sekijima (Shinshu University Hospital), Kunihiro Yoshida (Shinshu University Hospital), Sadayuki Matsumoto (Tazuke Kofukai, Medical Research Institute, Kitano Hospital), Toshihiko Suenaga (Tenri Hospital), Hiroshi Yaguchi (The Jikei University Kashiwa Hospital), Akira Kurita (The Jikei University Kashiwa Hospital), Masahiko Suzuki (The Jikei University Katsushika Medical Center), Masashi Hamada (The University of Tokyo Hospital), Shoji Tsuji (The University of Tokyo Hospital), Ryuji Sakakibara (Toho University Sakura Medical Center), Masaaki Kato (Tohoku University Hospital), Hitoshi Warita (Tohoku University Hospital), Kentaro Tokuoka (Tokai University Hachioji Hospital), Eiichiro Nagata (Tokai University Hospital), Yoshimichi Miyazaki (Tokushima University Hospital), Koji Fujita (Tokushima University Hospital), Wataru Sako (Tokushima University Hospital), Takanori Yokota (Tokyo Medical And Dental University, Medical Hospital), Hidehiro Mizusawa (Tokyo Medical And Dental University, Medical Hospital), Kazunori Nanri (Tokyo Medical University Hachioji Medical Center), Hiroo Terashi (Tokyo Medical University Hachioji Medical Center), Hitoshi Aizawa (Tokyo Medical University Hospital), Akihiro Kawata (Tokyo Metropolitan Neurological Hospital), Eiji Isozaki (Tokyo Metropolitan Neurological Hospital), Kenji Nakashima (Tottori University Hospital), Akira Tamaoka (University of Tsukuba Hospital), Yoshihisa Takiyama (University of Yamanashi Hospital), Toru Kawanami (Yamagata University Hospital), Fumiaki Tanaka (Yokohama City University Hospital).
Contributors MN, OO and YS contributed to the concept or design of the study. MY analysed the study data. AH advised the study data analysis. All authors contributed to the data interpretation and writing of the manuscript and approved the final version of the manuscript.
Funding Kissei Pharmaceutical funded the studies and was involved in designing the study, data collection, data analysis, data interpretation and writing of the report. The corresponding author had full access to all the data in the studies and had final responsibility for the decision to submit for publication.
Competing interests MN has received grants and personal fees from Kissei Pharmaceutical during the conduct of the study; and has received grants and personal fees from Astellas Pharma, Daiichi-Sankyo, Sumitomo Dainippon, Eisai, FP, GlaxoSmithKline, Japan Blood Products Organization, Kyowa Hakko Kirin, Mitsubishi Tanabe, MSD, Novartis, Otsuka, Pfizer, Sanofi and Takeda, outside the submitted work. OO has received grants and personal fees from Kissei Pharmaceutical during the conduct of the study; and has received grants and personal fees from Astellas Pharma, Boehringer Ingelheim, Daiichi-Sankyo, Eisai, Eli Lilly, FP, GlaxoSmithKline, Japan Blood Products Organization, Kyowa Hakko Kirin, Mitsubishi Tanabe, MSD, Novartis, Otsuka, Pfizer, Sanofi, Shionogi, Sumitomo Dainippon and Takeda, outside the submitted work. AH has received personal fees from Kissei Pharmaceutical during the conduct of the study. YS and MY are employees of Kissei Pharmaceutical. YS has a patent P2017-14198A issued.
Patient consent for publication Not required.
Ethics approval These studies were implemented based on ethical policies expressed in Good Clinical Practice (GCP) and the Declaration of Helsinki. Implementation of these studies was approved by the institutional review board of each facility prior to the start of the studies. Written informed consent was obtained from each patient.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available. Additional data analyses are acceptable on reasonable request.