Article Text

Abstract
Primary lateral sclerosis (PLS) is a neurodegenerative disorder of the adult motor system. Characterised by a slowly progressive upper motor neuron syndrome, the diagnosis is clinical, after exclusion of structural, neurodegenerative and metabolic mimics. Differentiation of PLS from upper motor neuron-predominant forms of amyotrophic lateral sclerosis remains a significant challenge in the early symptomatic phase of both disorders, with ongoing debate as to whether they form a clinical and histopathological continuum. Current diagnostic criteria for PLS may be a barrier to therapeutic development, requiring long delays between symptom onset and formal diagnosis. While new technologies sensitive to both upper and lower motor neuron involvement may ultimately resolve controversies in the diagnosis of PLS, we present updated consensus diagnostic criteria with the aim of reducing diagnostic delay, optimising therapeutic trial design and catalysing the development of disease-modifying therapy.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Primary lateral sclerosis (PLS) is a characteristically slowly progressive and selective neurodegenerative disorder primarily affecting the adult central motor system. Progressive muscle stiffness leads to an insidious loss of mobility typically with the development of corticobulbar dysfunction, which may be the initial symptom for a minority. Diagnostic criteria for PLS proposed 75 years ago recognised the potential for clinical overlap in the early symptomatic phase with the more common disorder amyotrophic lateral sclerosis (ALS).1 Like PLS, upper motor neuron (UMN)-predominant ALS has a significantly slower rate of progression compared with classical forms of ALS, with survival frequently extending into a second decade from onset of symptoms.2 The development of clinically obvious and functionally significant, progressive lower motor neuron (LMN) involvement is inevitable in ALS, in contrast to PLS, but may not emerge for several years from the initial clinical UMN syndrome.3 As a result, criteria for the definite diagnosis of PLS have enshrined a minimum duration of symptoms, varying from 3 to 5 years.1 4 5
Among the earliest reported cases, many of those that were said to have a hereditary component1 would now be recognised within the spectrum of hereditary spastic paraplegia (HSP). The development of non-invasive neuroimaging has brought further structural, inflammatory and metabolic mimic disorders into consideration (see later). A ‘gold standard’ postmortem histopathological signature for PLS has proved elusive. While neuronal and glial cytoplasmic inclusions of the 43 kDa transactive response DNA-binding protein, TDP-43, are common to 97% of cases of ALS (across disparate monogenetic and apparently sporadic cases), there have been very few postmortem studies of PLS in the modern era of immunohistochemistry.6 7 Debate as to whether PLS represents an extreme end of a continuum with ALS, or a distinct disorder is ongoing.
The clinical imprecision in the diagnosis, along with some uncertainty about overlap with UMN-predominant ALS has become an obstacle to therapeutic development for PLS. As the result of a meeting of international PLS experts (3 May 2019, Philadelphia, Pennsylvania, USA), a working group set forth to create more permissive diagnostic criteria, in an effort to spur therapeutic development and to accelerate research into the basic histopathology of PLS.
The core clinical syndrome
There have been consistent clinical observations reported across multiple case series in PLS.8 Mean age at symptom onset is around 50 years which is at least a decade earlier than non-familial ALS, and a decade later than HSP. While there have been cases reported with symptoms beginning in childhood, many of those might now be linked to developmental or monogenetically mediated disorders. A male predominance has been consistently noted in PLS (range 2–4:1).
An insidious onset is the rule in PLS, so that individuals are unlikely to reach specialised neurological services soon after the very earliest symptoms. For the majority of patients, symptoms emerge in the lower limbs first, but for a significant minority in the corticobulbar pathways with dysarthria and often prominent emotionality (pseudobulbar affect). Although dysphagia may become marked, the value of gastrostomy is far less clear than in ALS, and the need for non-invasive ventilation in PLS more exceptional. Lower limb involvement in the early symptomatic phase may be articulated as a sense of dysequilibrium or loss of fluidity in gait. Prominent sensory involvement should not be evident. Spasticity with pathological hyperreflexia are invariable examination findings. Although PLS typically generalises to include the upper limbs, a focal upper limb onset to symptoms is very unusual in PLS.9
Additional clinical features
Bladder instability resulting in frequency and varying degrees of retention is a common accompaniment to PLS. The presence of wider brain involvement in PLS is increasingly recognised clinically but is typically minor in relation to the core clinical syndrome. Extrapyramidal features are occasionally observed.10 11 Although significant cognitive impairment within the spectrum of frontotemporal dementia (FTD) has been documented,3 12–14 the frequency of FTD is significantly lower than in ALS. While the initial lower limb symptom onset in PLS is commonly symmetrical, a progressive hemiplegia is a very rare phenotype originally described eponymously by Mills.15 Such cases need a particularly careful search for focal lesions as they may mimic ‘solitary sclerosis’.16 Once these have been excluded, cases of slowly progressive hemiplegia, typically with bilateral UMN signs despite unilateral weakness,17 are currently reasonable to consider within the spectrum of PLS.
Consensus diagnostic criteria
A revised framework is proposed to facilitate the earlier diagnosis of PLS (box 1). The choice of a minimum age of 25 years at point of diagnosis (‘probable’ or ‘definite’ PLS) reflects a pragmatic decision to minimise the risk of highly atypical cases distorting outcome in future therapeutic trials.
Consensus diagnostic criteria for primary lateral sclerosis (PLS)
The diagnosis of PLS requires:
the presence of:
age ≥ 25 years;
symptoms of progressive upper motor neuron (UMN) dysfunction for at least 2 years;
signs of UMN dysfunction* in at least two of three regions: lower extremity, upper extremity, bulbar.
the absence of:
sensory symptoms (unexplained by comorbid condition);
active lower motor neuron (LMN) degeneration†;
alternative diagnosis‡: UMN pathology demonstrated on neuroimaging, or identified through biofluid testing that provides a plausible alternative explanation for the clinical syndrome.
2. Diagnostic certainty
Probable PLS is defined by the absence of significant active LMN degeneration 2–4 years from symptom onset.
Definite PLS is defined by the absence of significant active LMN degeneration 4 or more years from symptom onset.
*Clinical signs, including spasticity and associated weakness, pathological hyperreflexia (including Hoffman’s sign and bilateral extensor toe responses), pseudobulbar affect. Laboratory evidence of UMN dysfunction from emerging neuroimaging, neurophysiological and neurochemical biomarkers (see ‘Emerging technology’ section) is pending validation.
†Minimally increased insertional activity and positive sharp waves or fibrillation potentials in extremity muscles are permitted (see ‘Electromyographic considerations’ section).
‡see ‘Differential diagnosis’ section.box 2
Electromyographic considerations
The late development of LMN involvement in some cases of UMN-predominant ALS has the potential to lead to misclassification of PLS. This issue is complicated by the presence of ‘low-grade’, non-progressive electromyographic (EMG) signs of limited muscle denervation in some cases of PLS.4 18–23
Pringle et al allowed ‘at most, occasional fibrillation and increased insertional activity in a few muscles (late and minor)’.4 Gordon e t al divided patients into pure PLS and UMN-dominant ALS, with 13 of 29 patients with pure UMN developing EMG denervation and LMN signs on average between 3 and 4 years from symptom onset, four of who met criteria for ALS.5 Other series have not shown such a definitive transition to UMN-predominant ALS, nor the development of LMN signs on examination, though some have reported a latency exceeding 4 years.3
Singer et al divided patients into two categories, with and without EMG findings.20 Neither group went on to develop ALS. They found the group with ‘evidence of active denervation potentials (increased insertional activity, fibrillations and/or positive sharp waves) in one or more muscles’ were older and progressed more rapidly. None of the other series reported any significance to minor EMG findings. Mitsumoto et al included patients with normal EMG but allowed minimal changes in one muscle.21 Fournier et al identified 217 patients with pure UMN disease at 20 clinic sites and divided the groups into normal EMG, and minor denervation, and found no differences between groups.22
In general, most patients with minor denervation in a rare extremity muscle remain a pure UMN syndrome, so that these minimal findings on EMG have been permitted. The inverse, someone with minor EMG findings in a rare muscle initially who at 4 years have a completely normal EMG, would also support a PLS diagnosis. Notwithstanding the limitations of using EMG as a biomarker for LMN involvement, the recognition of a pragmatic category of ‘probable PLS’ for those with a progressive, idiopathic upper motor syndrome of between 2 and 4 years from symptom onset, reflects a desire to facilitate earlier inclusion of patients with PLS in future trials of potentially disease-modifying therapy before disability becomes advanced.
Differential diagnosis
HSP has the most clinical overlap with the early symptomatic phase of lower limb-onset PLS.24 HSP and PLS are both fundamentally clinical syndromes. A significant proportion of individuals confidently labelled as HSP on clinical grounds will not carry a recognised pathological genetic variant. Analysis of 90 patients with apparently sporadic UMN syndrome with phenotypes of HSP (involvement of legs only), HSP-PLS overlap (involvement of arms and legs) and PLS (bulbar involvement) showed significant overlap in the age of symptom onset and no differences between the groups in features classically used to distinguish the two, such as mild dorsal column dysfunction or urinary urgency.25
Mimic disorders for PLS are rare and high-resolution clinical MRI of the brain and spinal cord will eliminate the majority of these. With the exception of the neurodegenerative category, the plausibility of many alternative diagnoses greatly diminishes with the duration of a progressive pure UMN syndrome at the time of clinical assessment (table 1).
Differential diagnosis of primary lateral sclerosis
The role of genetic testing
The classical syndrome of PLS appears to be sporadic and the diagnosis based on clinical features. Screening of panels for pathogenic genetic variants associated with spastic paraparesis (eg, SPAST) is warranted in cases of progressive UMN syndromes restricted to symmetrical lower limb involvement. It is reasonable to routinely exclude the most common hereditary cause of ALS in Caucasian populations, namely an expansion in C9orf72 which may present with an UMN-predominant phenotype. However, the plethora of very rare genetic variants reported in association with pedigrees containing ALS-like syndromes, including some with apparently pure UMN phenotypes, should not be considered routine tests in the diagnosis of PLS (box 2).
Genetic variants reported in a small percentage of upper motor neuron-predominant syndromes
SPG7*
ALS2*
D4S2963*
C9orf72
DCTN1
PARK2
ERLIN2
FIG4
SYNE2
VEGFA
CLN6
BTD
LRKK2
SQSTM1*
KIF5a*
KIF1a
Primary lateral sclerosis appears to be a sporadic disorder essentially, with diagnosis based primarily on clinical features rather than genotype.
*Familial cases.
Emerging technology
A range of neuroimaging and neurophysiological tools have clear potential to quantify the UMN lesion and may help to refine the diagnostic pathway for PLS.26 Conversely, tools aimed at demonstrating subclinical LMN involvement, such as muscle ultrasound for the detection of fasciculation,27–29 may ultimately offer additional value for the earlier distinction from UMN-predominant ALS.
Transcranial magnetic stimulation studies have noted greater central motor conduction times in PLS compared with ALS,18 in addition to high threshold measures for cortical stimulation, leading to the development of relative cortical inexcitability, a feature that reliably distinguishes PLS from HSP.30 Focus on beta-band EMG has considered intermuscular coherence as a potential distinguisher of PLS from ALS,31 and magnetoencephalography offers a broader analysis of differences in corticomuscular coherence.32
Neurofilaments are an emerging biofluid biomarker reflecting the intensity of neuronal loss in a range of neurological disorders. Levels tend to be much lower in PLS compared with ALS,33 reflecting its much slower progression. Cerebrospinal fluid chitinases, thought to be macrophage-derived, may show a differential pattern of involvement in PLS compared with ALS,34 but further studies are needed to explore the key distinction of PLS from UMN-predominant ALS.
One of the features noted in some established cases of PLS is a focal ‘knife edge’ atrophy of the precentral gyrus,4 which is strikingly absent in even advanced cases of ALS. With the refinement of more automated volumetric MRI analysis, coupled to large normative databases, it may be possible to quantify the degree of focal motor cortical atrophy at the individual patient level and integrate this within the diagnostic certainty algorithm. Similarly, the presence of focal fluorodeoxyglucose hypometabolism in the same region—the ‘stripe sign’—has been associated with PLS,35 but has not been validated against the core differential diagnosis of UMN-predominant ALS. Diffusion tensor imaging is a development of standard MRI permitting assessment of the integrity of large white matter tracts through the surrogate marker of the directionality of water diffusion. This suggests greater white matter damage in the region of the central corpus callosum in patients with PLS, but this currently lacks the sensitivity and specificity for the diagnosis of individual patients36–38 (figure 1). Cerebellar involvement,38 39 corticospinal tract fluid attenuated inversion recovery (FLAIR) hyperintensity40 and quantitative susceptibility mapping of iron deposition in the motor cortex41 have all been noted as increased in PLS. The continued development of volumetric spinal cord imaging and its integration with cerebral structural measures may offer greater potential for distinguishing PLS cases.42

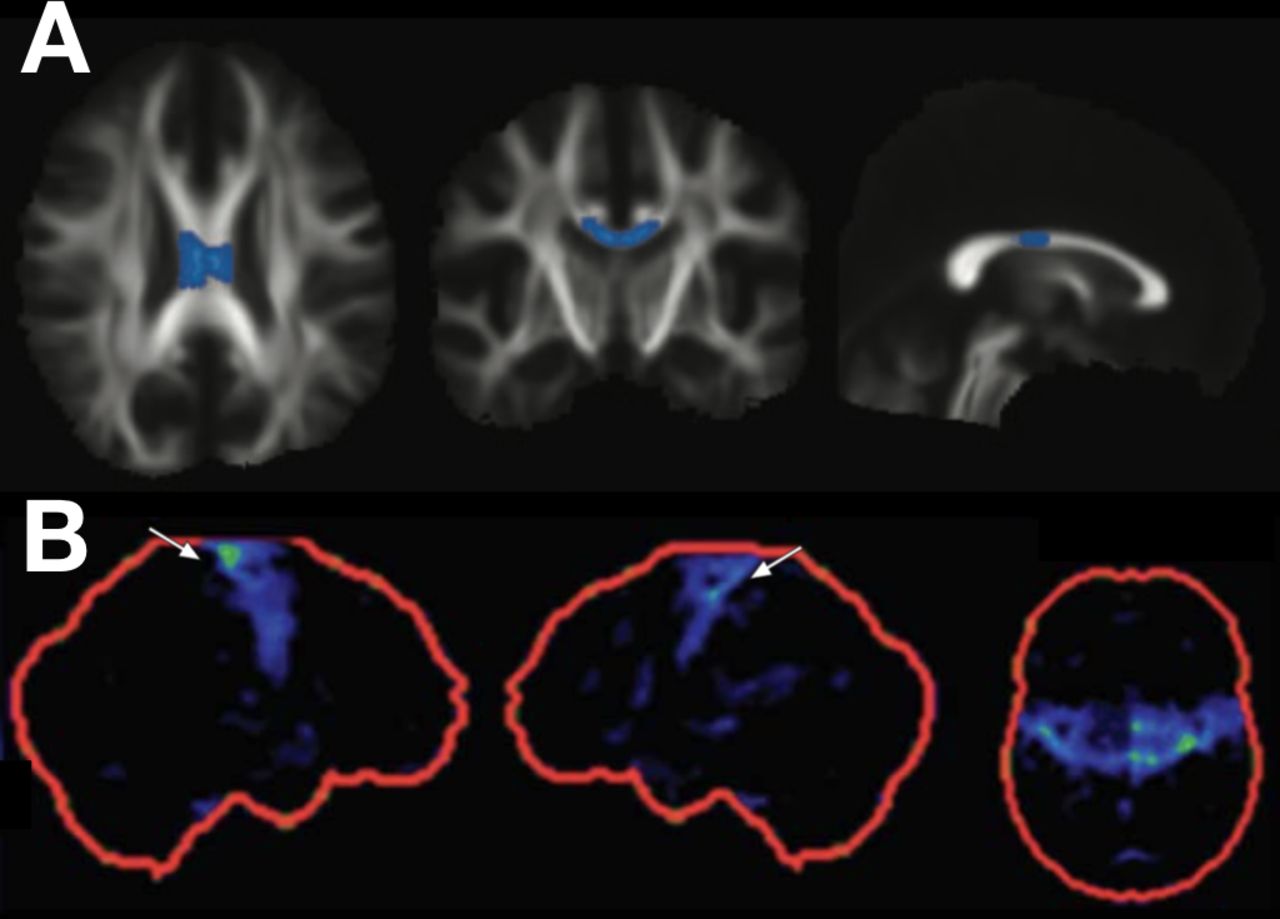{kind=link}
Diffusion tensor imaging and fluorodeoxyglucose (FDG) positron emission tomography (PET) findings in cases of primary lateral sclerosis (PLS). Mean diffusivity in the mid-portion of the corpus callosum is increased in cases of PLS compared with amyotrophic lateral sclerosis (row A, PLS vs ALS group findings highlighted on axial, coronal and sagittal views of the white matter tract skeleton; with kind permission of the author, see Iwata et al 37). Focal hypometabolism may be seen in the primary motor cortices in PLS (row B, FDG PET z-score images in the right sagittal, left sagittal and axial planes; with kind permission of the author, see Claassen et al 35). Neither of these techniques yet has sufficient sensitivity or specificity to be applied in isolation for the diagnosis of PLS.
All of these tools require further prospective studies to define their precise value in the diagnostic algorithm for PLS.
Conclusions
Developments in neuroimaging, neurophysiology and molecular biology have not diminished the long-standing recognition of PLS as a distinct, clinical ly-defined syndrome. Its rarity and prolonged survival trajectory have resulted in a degree of neglect in terms of therapeutic trials compared with ALS. Traditional reliance on EMG with overinterpretation of minimal markers of LMN involvement may contribute to significant diagnostic delay. The development of an international registry that includes all those with ‘probable PLS’ will allow a more precise delineation of the pathogenesis from ALS, and accelerate therapeutic developments.
Beyond primary disease-modifying therapy, the most pressing unmet need for patients with PLS may well be the treatment of core symptoms rather than extension of survival. The development of more effective relief of spasticity that does not sacrifice muscle strength would have a great impact for those living with PLS, notwithstanding the long-term desire for neuroprotective or regenerative therapy. Recognising that advances in molecular phenotyping may well supersede purely clinical diagnostics, it is hoped that these pragmatic criteria will provide greater confidence to reduce diagnostic delay, thus allowing access to potentially disease-modifying therapies at lower levels of disability.
References
Footnotes
Twitter @jnnp_bmj
Collaborators The following delegates of the 2nd International PLS Conference (3 May 2019, Pennsylvania, USA) contributed to these consensus criteria: Senda Ajroud-Driss, Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA. Jinsy A. Andrews, Director of Neuromuscular Clinical Trials, Columbia University Irving Medical Center, New York, NY, USA. Suma Babu, Sean M. Healey & AMG Center for ALS, Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA. Peter Bede, Associate Professor, Computational Neuroimaging Group, Trinity College Dublin, Ireland. Michael Benatar, Professor of Neurology and Public Health Sciences, Chief, Neuromuscular Division Executive Director, The ALS Center, Department of Neurology, Miami, FL, USA. Sheena Chew, Massachusetts General Hospital, Boston, MA, USA. Robin Conwit, Program Director, Division of Clinical Research, NINDS, NIH, Bethesda, MD, USA. Merit Cudkowicz, Neurologist, Clinical Researcher, Professor of Neurology, Chief, Neurology Department, Harvard Medical School, Massachusetts General Hospital, Boston, MA, USA. Mamede de Carvalho, Faculdade de Medicina-IMM, Universidade de Lisboa, Department of Neurosciences, Hospital de Santa Maria-CHULN, Lisbon, Portugal. Vivian E. Drory, Head of Neuromuscular Diseases Unit, Department of Neurology, Tel-Aviv Sourasky Medical Center, Tel-Aviv, Israel. Lauren Elman, Associate Professor of Neurology, University of Pennsylvania Medical Center, Philadelphia, PA, USA. J. Americo M. Fernandes, Associate Professor, University of Nebraska Medical Center, Omaha, NE, USA. Dominic Ferrey, Department of Neurosciences, University of California at San Diego, La Jolla, CA, USA. Eoin Finegan, Computational Neuroimaging Group, Biomedical Sciences Institute, Trinity College Dublin, Ireland. Mary Kay Floeter, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA. Christina N. Fournier, Department of Neurology, Emory University, Atlanta, GA, USA. Raghav Govindarajan, Associate Professor, University of Missouri, Columbia, MO, USA. Volkan Granit, Department of Neurology, Neuromuscular Division, University of Miami, Miller School of Medicine, Miami, FL, USA. Orla Hardiman, Clinical Professor Neurology, Consultant Neurologist at the National Neuroscience Centre, Director of the National ALS Clinic & Irish ALS Research Group, Trinity Institute of Neuroscience, Trinity College Dublin, Ireland. Ghazala Hayat, Professor of Neurology, St. Louis University, St. Louis, MO, USA. Terry D. Heiman-Patterson, Temple University Lewis Katz School of Medicine, Philadelphia, PA, USA. Annemarie Hübers, Service de Neurologie, Hôpitaux Universitaires de Genève, Switzerland. Edward D. Huey, Associate Professor of Psychiatry and Neurology, Taub Institute for Research on Alzheimer's Disease and the Aging Brain, Columbia University Irving Medical Center, New York, NY, USA. Omar Jawdat, Assistant Professor of Neurology, Director, ALS Clinic, University of Kansas Medical Center, Kansas City, KS, USA. Osamu Kano, Professor and Chairman, Division of Neurology, Department of Internal Medicine, Toho University Faculty of Medicine, Tokyo, Japan. Yaz Y. Kisanuki, Department of Neurology, The Ohio State University Wexner Medical Center, Columbus, OH, USA. Dale Lange, Chair, Department of Neurology; Neurologist-in-Chief, Hospital for Special Surgery; Professor of Neurology
Weill Cornell Medicine; New York, NY, USA. Ian Mackenzie, Professor, Neuropathology, Department of Pathology and Laboratory Medicine University of British Columbia, Vancouver, Canada. David Marren, Spastic Paraplegia Foundation Mitsuya Morita Associate Professor, Chief of Rehabilitation Center, Jichi Medical University, Tochigi, Japan. Jennifer Murphy, University of California, San Francisco, CA, USA. Sharon Nations, Professor, Department of Neurology and Neurotherapeutics, University of Texas Medical Center, Dallas, TX, USA. Björn Oskarsson, Associate Professor of Neurology, Jacksonville, Director ALS Center of Excellence, Mayo Clinic, Jacksonville, FL, USA. Sabrina Paganoni, Healey Center for ALS, Massachusetts General Hospital, Boston, MA, USA. Erik P. Pioro, Director, Section of ALS & Related Disorders, Professor of Neurology Department of Neurology, Cleveland Clinic, Cleveland, OH, USA. Seth L. Pullman, Columbia University, NY, USA. Kourosh Rezania, Department of Neurology, University of Chicago, Illinois, USA. Guy Rouleau, Director, Montreal Neurological Institute and Hospital, Chair, Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada. Stephen Scelsa, Site Medical Director, Department of Neurology, Mount Sinai Downtown and Mount Sinai Beth Israel; Director of the ALS Center, Mount Sinai Downtown Union Square; Professor of Neurology, the Icahn School of Medicine, New York, NY, USA. Nailah Siddique, Neurogenomics Lab, Northwestern University Feinberg School of Medicine, Chicago, IL, USA. Juan Solano, Neuromuscular Disorders Research Group, Universidad de Antioquia, Medellín, Colombia Marka van Blitterswijk Department of Neuroscience, Mayo Clinic, Jacksonville, FL, USA. David Walk, Professor, Department of Neurology, University of Minnesota, Minneapolis, MN, USA. James Wymer, University of Florida, Gainesville, FL, USA. Cuiping Zhao, Department of Neurology, Qilu Hospital, Shandong University, China.
Contributors MRT drafted the manuscript, revised and synthesised the final version and is the guarantor. RJB co-drafted the EMG section and provided intellectual content and revisions to the overall manuscript. PC drafted table 2 and provided intellectual content and revisions to the overall manuscript. JKF provided intellectual content and revisions to the overall manuscript. MBH provided intellectual content and revisions to the overall manuscript. MCK provided intellectual content and revisions to the overall manuscript. JR provided intellectual content and revisions to the overall manuscript. VS drafted table 3 and provided intellectual content and revisions to the overall manuscript. ZS provided intellectual content and revisions to the overall manuscript. JS co-drafted the EMG section and provided intellectual content and revisions to the overall manuscript. LHvdB provided intellectual content and revisions to the overall manuscript. HM conceived the article and provided intellectual content and revisions to the overall manuscript.
Funding MRT is supported by the Motor Neurone Disease Association. MCK was supported by the National Health and Medical Research Council of Australia Program Grant (#1132524), Partnership Project (1153439) and Practitioner Fellowship (1156093). VS was supported by the Italian Ministry of Health, AriSLA (Fondazione Italiana di Ricerca per la SLA), and E-Rare Joint Transnational Call. ZS received funding from Cytokinetics, Biohaven & Biogen. HM received funding from CDC, MDA, SPF, Cytokinetics and Tsumura.
Competing interests MRT is on the Scientific Advisory Board for Orphazyme, has been a paid consultant for Genentech Inc. (2017) and anonymous clients separately through GLG Consulting on the topic of ALS diagnosis, management and biomarker development. He received neurofilament assay kits in-kind from Euroimmun UK (2018).PC has reveived honoraria from Biogen & Cytokinetics. ZS has received honoraria from Wiley, Cytokinetics & Biohaven. HM is on the Advisory Board of Mitsubishi-Tanabe & Biohaven.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.