Article Text

Abstract
Background A mutation in C9orf72 constitute a cross-link between amyotrophic lateral sclerosis (ALS) and fronto-temporal dementia (FTD). At clinical manifestation, both patient groups may present with either cognitive impairment of predominantly behaviour or language (in FTD) or motor dysfunctions (in ALS).
Methods In total, 36 non-symptomatic mutation carriers from ALS or FTD families were examined, including 21 subjects with C9orf72 and 15 with SOD1 mutations. Data were compared with 91 age-matched, education-matched and gender-matched healthy subjects (56 were first-degree relatives from ALS or FTD families, 35 with no known family history of ALS/FTD). MRI scanning for diffusion tensor imaging was performed to map fractional anisotropy (FA). Subjects performed an extensive neuropsychological assessment to address verbal fluency, language, executive, memory and visuospatial function. Measurements were repeated after 12 months.
Results C9orf72 expansion carriers performed significantly worse in verbal fluency and non-verbal memory and presented with distinct alterations in structural white matter integrity indicated by lower FA values in inferior and orbitofrontal cortical areas compared with carriers of SOD1 mutations or healthy subjects. Loss of structural integrity was associated with decreased verbal fluency performance. White matter alterations and cognitive performance showed no changes over 12 months in all subjects.
Discussion Reduced verbal fluency performance seems to be a distinct clinical feature of C9orf72 carriers before symptomatic disease onset without evidence for change over time in our cohort. The results support the emerging hypothesis of a general disorder in development in addition to neurodegeneration in C9orf72 carriers.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Clinical manifestation is motor predominant in amyotrophic lateral sclerosis (ALS), whereas fronto-temporal dementia (FTD) presents primarily with alterations of behaviour and cognition. Despite these disparities, both entities share some common hallmarks of cognitive impairment (including behaviour or language changes)1 and a hexanucleotide GGGGCC-repeat expansion in C9orf72 (HRE) as the most prevalent genetic cause of ALS and FTD in Caucasian populations.2 In families carrying C9orf72HRE, different clinical phenotypes may cosegregate in the form of either ALS, FTD or FTD–ALS of mostly behavioural subtype. For FTD, cognitive impairments are part of the clinical phenotype but also ALS patient groups may present with variable degree of cognitive impairment when clinical motor symptoms become apparent. Minor cognitive deficits have been reported in about 50% of ALS patients, with 30% reaching a clinically relevant threshold (defined as below threshold within more than one cognitive domain),3 while only 5%–10% present with a full-blown ALS–FTD phenotype. The cognitive domains being most prominently affected in ALS are verbal fluency, language, social cognition, executive functions and memory.3 Cognitive function deficits may be associated with white matter changes in frontal areas of ALS patients;1 4 Mioshi and colleagues5 provided evidence that cognitive changes in newly diagnosed ALS patients occur early in the disease course and may even precede motor impairment. In FTD, first cognitive–behavioural alterations may emerge many years before clinical diagnosis.1 5 However, the time of onset and evolution of these cognitive symptoms is unclear. In earlier studies in C9orf72 mutation carriers, structural and functional changes on cortical level may precede the emergence of clinically relevant symptoms of ALS6 and FTD by a median of 12.5 years7 and possibly up to three decades8 before expected overt symptomatic onset. Studies provided evidence that C9orf72 carriers show lower scores in letter fluency or processing speed compared with healthy subjects which was associated with reduced cortical volume.9 Executive functions and language skills such as naming may show subtle changes up to 5 years before expected onset.10
The association between C9orf72 mutations and structural and functional changes in the central nervous system (CNS) may be mechanistically explained by the C9orf72 protein being believed to play a key role in the development of the CNS11 and in particular in synaptic structures.12 Also, C9orf72-linked accumulation of TDP43 in specific brain regions in ALS is critical during neuronal development and cell death probably following complex disturbances of nuclear and cytoplasmic functions also in the preclinical state.13–15
Presently, the link of C9orf72 to clinical changes of development is missing. We are longitudinally following a cohort of apparently asymptomatic individuals who are relatives of patients with mutations in ALS–FTD causing genes. This pre-ALS study includes regular biomarker, imaging, clinical and neuropsychological testing performed unknown to genotype of the study subject. We here addressed the hypothesis that developmental changes may be observed in ‘asymptomatic’ carriers of C9ORF72 compared with other mutation carriers and healthy subjects.
Methods
Study cohorts
In total, n=127 participants were included. Of these, n=92 were from families with at least one member diagnosed with ALS (family member FM) according to the revised El Escorial criteria who was tested positive for a pathogenic mutation for familial ALS (fALS). At least one of the affected FM had attended our clinic before (index patient). All FMs of this index patient were contacted and invited to take part in the study. In case of interest, they contacted our clinics and underwent a series of tests, including MRI and neuropsychological testing (the FM group).
None of the subjects had a family history of any neurological disorder apart from ALS and/or FTD (eg, genetic AD). Further, none of the subjects had subjective or clinical manifestation of ALS or FTD. All subjects were tested for the most common genes known to cause fALS. Of the n=92 FM subjects, n=22 carried hexanucleotide repeat expansion mutations in the C9orf72 gene (C9orf72HRE), n=15 SOD1 mutations and n=56 no mutation known to be involved in the pathogenesis of ALS or FTD (FM control). The remaining nine subjects had mutations in rare ALS genes (n=3 KIF5A; n=3 FUS/FUS+TBK1; n=1 NEK1; n=1 SETX; n=1 TDP43), and due to low sample size, they were excluded from statistical analysis. The genetic analysis was performed according to state-of-the art principles16 (details available on request). Southern blot was performed to determine the repeat length for C9orf72HRE (between 70 and 2600 repeats). The asymptomatic participants were not informed about their genotypes but had received genetic counselling and knew that they might be at risk of being a mutation carrier and at risk of later developing ALS or FTD.
For each FM subject, suspected time until expected onset was calculated using the age of onset of the index patient minus the current age of the participant6 10 (for details, see table 1). Additionally, n=35 controls with no evidence for family history of ALS or FTD and without mutation in an fALS gene were included (no-FM control). They were matched for age, sex and education to the fALS group (table 1).
Demographics of subjects with either a C9orf72 mutation or SOD1 mutation from families with members diagnosed with ALS or FTD (family member, FM)
Supplemental material
Participants with motor or cognitive signs indicating ALS/FTD or with insufficient knowledge of German language were excluded. History of or current comorbid neuropsychiatric disorder was recorded but did not serve as an exclusion criterion. n=29 (22%) subjects reported on a history of clinically relevant lifetime depression or other lifetime psychiatric disorders (anxiety disorder, compulsive behaviour disorder, schizophrenia and psychosis).
Study design
Procedures
The MRI scan was performed first followed (mean latency up to 2 hours) by neuropsychological testing. Standardised and semistructured interviews demographics, cognitive and behavioural changes and changes in affective state were recorded.
Longitudinal analysis
A subsample of n=25 subjects was tested again after 43±13 months. There was no statistical significant difference between those subjects that received an additional scanning compared with those who did not with regard to age, gender, education or predicted years until onset (all with F>1.86/χ²=0.61 with p>0.05). Drop-out rate was due to organisational reasons (time restriction, inability to attend the clinics a second time). All subjects for second scanning received complete neuropsychological testing and MRI scanning, following the same protocol as for the first measurement.
Neuropsychological assessment
Depressiveness and anxiety was evaluated using the Hospital Anxiety and Depression Scale (HADS).17 For cognitive screening and to exclude major cognitive deficits, the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) was used18 with language-specific stratified cut-off scores.19 The ECAS includes an ALS specific section encompassing executive functions including social cognition and verbal fluency, and language function. Furthermore, there is a non-ALS specific section focussing on memory and visuospatial functions. Age and education stratified cut-offs were used but none of the participants was below threshold. A carer behaviour screen of the ECAS addressing five domains characteristic of FTD and three domains for psychotic changes was included. n=116 primary caregivers (all first-degree relatives) evaluated changes in behaviour. None of the participants provided clinically relevant signs of FTD.
Additionally, all participants were tested on different cognitive functions using the following standard instruments: executive function of verbal fluency (phonematic fluency ‘S’ and ‘M’; Regensburger Wortflüssigkeitstest)20 and design fluency,21 verbal (Paarassoziationslernen)22 and non-verbal memory (Doors test),23 and attention and processing speed (Trail Making Test).24
Statistics on cognitive performance
The statistical analysis was performed with Statistical Package for the Social Sciences V.25.0. To test for normal distribution, Kolmogorov-Smirnow-Test was performed and according to normal distribution, parametric tests were used. For group differences in cognitive performance, analysis of covariance with confounder anxiety and posthoc Scheffé was performed. Effect size is given as η2 with 0.01 indicating a small, 0.06 a medium and 0.14 a large effect according to Cohen.25 Further, regression analysis was performed to determine the impact of anxiety on cognitive performance. Pearson correlation was used for correlation analysis of cognitive performance and white matter changes and time until expected onset performance with effect size r (<0.3 no effect, 0.3–0.5 medium effect, >0.05 large effect). To find significance in small subject samples indicates rather robust results according to Cohen. For the follow-up measurement, repeated measure analysis of variance was used for all subjects and for C9orf72 carriers only with regard to all cognitive domains measured. A threshold of p<0.05 (two-tailed) was used for statistical interference.
MRI scanning and analysis
MRI scanning was performed according to a standardised protocol (for details see reference26). The standardised MRI scanning protocol on a 1.5 Tesla Magnetom Symphony (Siemens Medical) with a 12-channel head coil was as follows: 60 gradient directions (b=1000 s/mm²) and two b=0 gradient directions, 64 slices, 128×128 voxels in-plane, slice thickness 2.5 mm, in-plane voxel size 2.5 mm x 2.5 mm, echo time 102 ms, repetition time 8700 ms. The MRI acquisition protocol partially (depending on the condition of the subject) included T2 and FLAIR (fluid-attenuated inversion recovery) sequences. All data sets were inspected by an experienced neuroradiologist for confounding vascular, neuroinflammatory or incidental pathology.
The diffusion tensor imaging (DTI) analysis software Tensor Imaging and Fibre Tracking27 was used for postprocessing and statistical analysis; the algorithms have been previously described in detail.26 28 In brief: after correction for eddy-currents and a standardised quality control protocol, stereotaxic normalisation to the Montreal Neurological Institute (MNI) space was performed iteratively using study-specific templates. To map white matter microstructure, from the stereotaxically normalised DTI, data sets of all subjects fractional anisotropy (FA) maps were calculated. A Gaussian filter of 8 mm full width at half maximum was applied for smoothing of FA maps for a good balance between sensitivity and specificity. Finally, FA maps were corrected for the covariate age. Statistical comparison by Student’s t-test was performed voxel-wise for FA values to detect changes between the subject groups (whole brain-based spatial statistics, WBSS). Voxels with FA values below 0.2 were not considered for statistical comparison, since cortical grey matter shows FA values up to 0.2. Statistical results were corrected for multiple comparisons using the false-discovery-rate (FDR) algorithm at p<0.05. Further reduction of the alpha error was performed by a spatial correlation algorithm that eliminated isolated voxels or small isolated groups of voxels in the size range of the smoothing kernel leading to a threshold cluster size of 256 voxels (for further information, see references26 28).
Correlation analysis (Pearson correlation) with clinical parameters was performed for averaged FA values from regions-of-interest (ROIs). The placement of the ROIs was data-driven initiated by the results of the WBSS. Spherical ROIs with a radius of 30 mm were placed and FA values within these ROIs were arithmetically averaged (FA values below 0.2 were not considered).
Results
Neuropsychology
Mood
All FM subjects with a family history of ALS were significantly more anxious (F=9.2, p=0.003) and more depressed (F=5.1, p=0.025) and reported a lower QoL (F=4.0, p=0.047; table 1) compared with healthy subjects without family history. There was no significant difference between groups with family history (subjects with either C9orf72 or SOD1 mutations) versus no mutations. Regression analysis showed that anxiety did not significantly explain variance in cognitive performance (for all cognitive subdomains <10% of variance with p>0.05).
Differences in cognition between groups
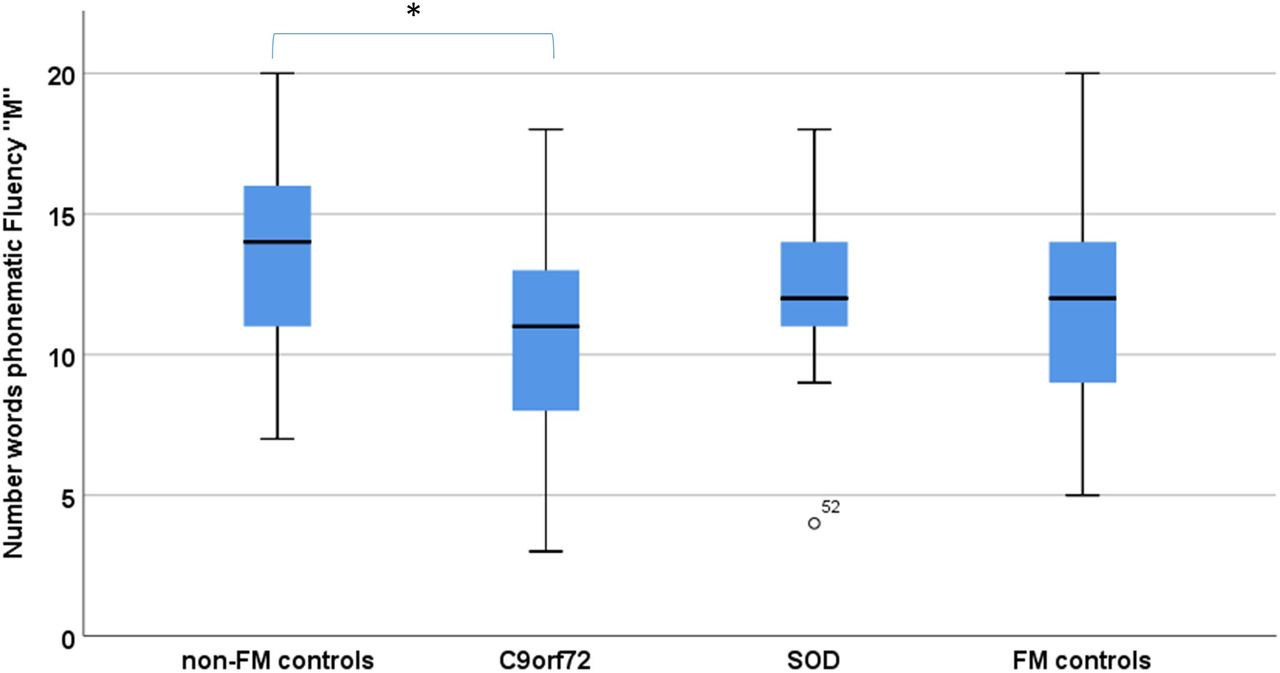
Groups differed significantly in phonematic verbal fluency (‘S’ and ‘M’) and visual memory (Doors B, table 1). In posthoc Scheffé comparison, C9orf72 carriers performed significantly worse in both phonematic fluency (‘M’ p=0.04, η2=0.07; figure 1) and visual memory (Doors B, p=0.04, η2=0.09) when compared with controls (no FM controls).
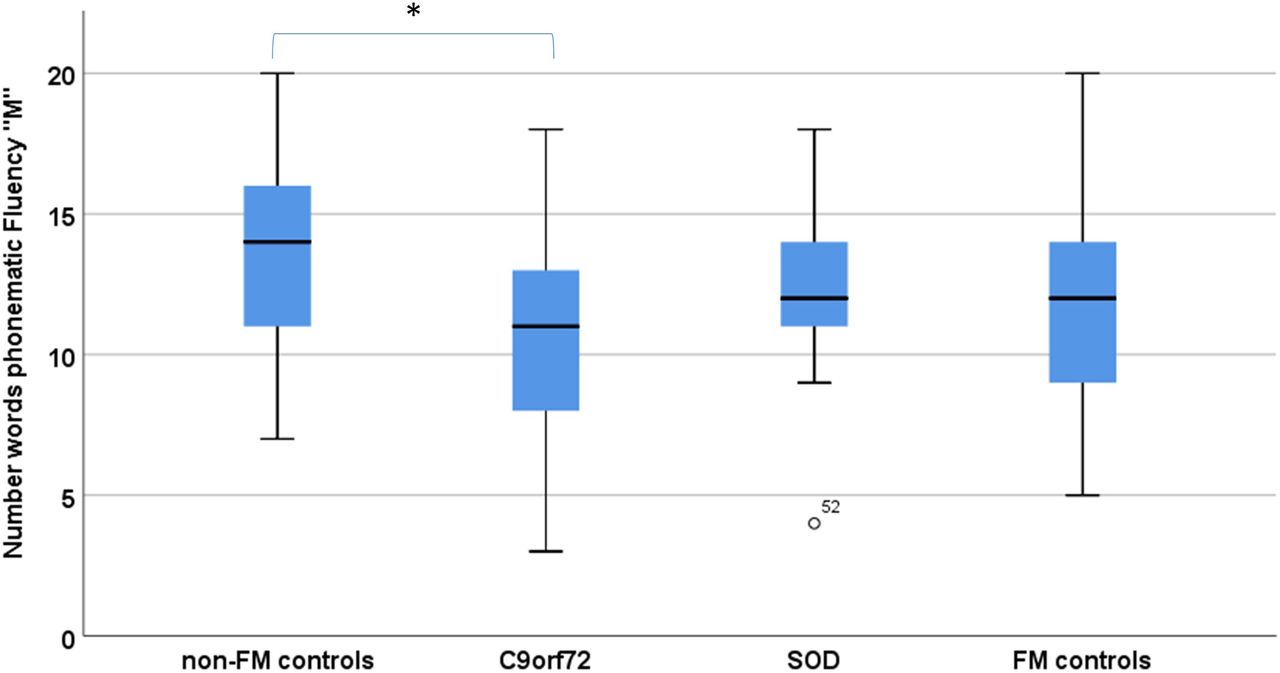
Verbal fluency performance in C9orf72 carriers (C9orf72), SOD1 mutation carriers (SOD1) compared with subjects with (FM controls) and subjects without a family history of ALS/motor neuron disease (non-FM controls). *Statistical significance with p<0.05. ALS, amyotrophic lateral sclerosis; FM, family member.
Both SOD1 mutation carriers and FM controls performed in the similar range like no FM controls. There was no significant difference in design fluency, verbal memory, and attention and processing speed for any group comparison. Also, ECAS overall score and ECAS subdomains (language, executive functions, restricted and free verbal fluency, verbal memory and visuospatial function) showed no significant differences between groups.
For follow-up measurements in C9orf72 carriers, there was no significant change in verbal fluency or visual memory nor any other cognitive domain measured. Also, the group of FM subjects showed no significant change in cognitive performance in the course of the study (all with r<0.3 and p>0.05).
Association of cognitive performance with demographics and clinical parameters
In C9orf72 carriers, visual memory performance and attention was associated with age, whereas none of the other cognitive functions including verbal fluency performance was associated with age or years until expected symptomatic onset.
Microstructural alterations
The WBSS comparison at the group level for the 22 C9orf72 mutation carriers versus 22 controls demonstrated bilateral regional alterations in white matter axonal structures at FDR-corrected p<0.05 projecting to the orbitofrontal cortex (left cluster size with peak at MNI −15/29/4, 5132 mm³, right cluster size with peak at MNI 12/26/0, 3393 mm³) and to the frontal cortex (cluster size with peak at MNI −15/18/35, 1225 mm³) (figure 2). These localisations were chosen as seed points for the consecutive ROI analysis. The choice of data-driven localisations is not a contradiction to previous studies29 given that the only significant alterations identified by WBSS were in the frontal regions. Averaged FA values from the ROI analysis (r=30 mm at MNI±19/31/3) were used for the correlation analysis to the neuropsychological parameters verbal fluency, DOORS and ECAS results, respectively.

White matter integrity analysis of n=22 asymptomatic C9orf72 carriers versus n=21 controls (non-FM controls). (A) Whole brain-based spatial statistics (WBSS) in slice wise view (MNI±19/31/3; near local bihemispherical maxima) (left) and in sagittal projectional view (right). (B) regions of interest (ROI)-based analysis. Left panel: ROI definition for group comparison and correlation analysis (r=30 mm at MNI±19/31/3). ROIs were displayed on a coloured FA-map background. Right panel: FA comparison for frontal ROIs at the group level for C9orf72 subjects versus controls. **p<0.001. FA, fractional anisotropy; FDR, false-discovery-rate; FM, family member; MNI, Montreal Neurological Institute.
Associations of cognitive performance and clinical and structural parameters
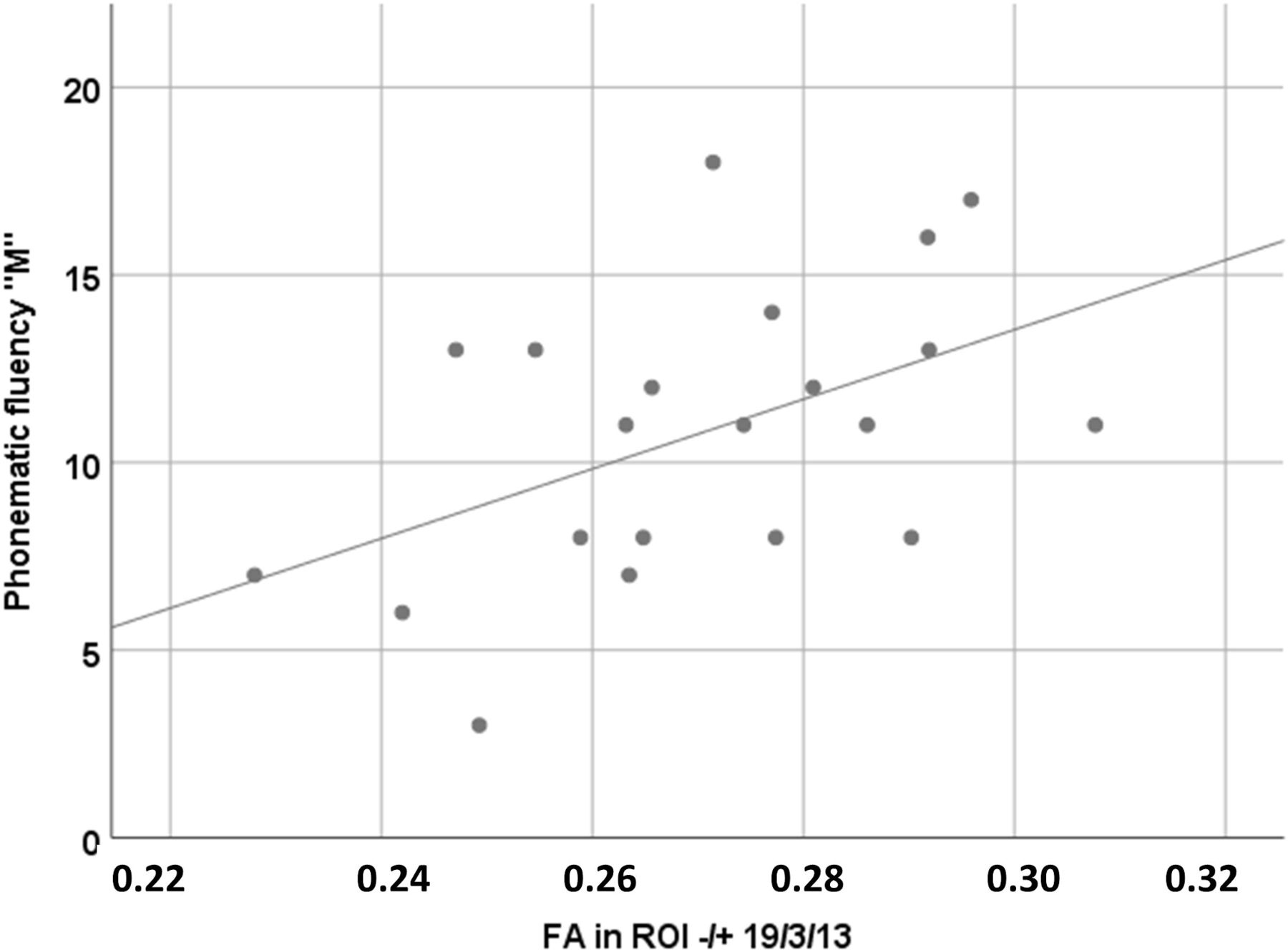
Loss of structural integrity in C9orf72 carriers was indicated by reduced FA values bilaterally in association fibres to the orbitofrontal cortex (figure 2) and was associated with performance in verbal fluency (r=0.47, p=0.03; figure 3), that is, the lower the FA values in association fibres to the bilateral inferior frontal cortex, the lower the cognitive performance in verbal fluency. White matter integrity was not associated with any of the other cognitive domains.
{kind=link}
{kind=link}
{kind=link}
Association of cognitive performance in verbal fluency and FA values in bilateral association fibres to the inferior frontal cortex (ROI ±19/3/13) in C9orf72 carriers; Pearson correlation indicated significant association (r=0.32, p=0.02). FA, fractional anisotropy.
Discussion
The aim of the study was to determine cortical substrates of preclinical cognitive alterations in C9orf72 carriers and to compare the results with other mutation carriers (SOD1) and subjects with no ALS causing mutations who were either from ALS families or not. Using whole brain-based DTI, presymptomatic C9orf72 carriers in this cohort presented with reduced FA values in inferior frontal and orbitofrontal areas. These areas are known to be affected by TDP43 pathology in ALS and FTD,13 14 in line with previous studies providing evidence for structural alterations in the clinical25 30 and in the preclinical state.4 7 8 C9orf72 carriers also performed significantly worse in the executive function of verbal fluency and non-verbal memory compared with subjects with SOD1 mutations or without any ALS causing genetic mutation. These changes are commonly observed in ALS patients3 and in mutation carriers in the preclinical state.4 These changes in our cohort were evident despite the absence of clinical signs and symptoms and were unrelated to age or expected time until disease onset in line with previous studies.6 This constellation suggests the presence of a general structural and cognitive deficit trait with currently no evidence for change over time. The higher the performance in verbal fluency, the higher were FA values indicative of intact WM tracts in those with good performance.
We know from literature that both impaired cognitive function31 and white matter alterations32 may be linked to early CNS development. Accordingly, it may be speculated that the cognitive and structural changes in C9orf72 carriers may not only be preclinical signs of ALS, but might be indicative of a general developmental disorder in C9orf72 carriers as suggested previously.33 Also, executive dysfunction and memory deficits are reported in subjects with delayed brain maturation.34–36 FA decreases in parallel with loss in cognitive performance throughout life,31 37 indicating a close association of structural integrity and cognitive performance. In the absence of clinical and radiological data from childhood we can't test this hypothesis, but our findings are consistent with this notion.
We have previously provided evidence for a similar association of structural and cognitive alterations in ALS patients.1 Yet, the concept in ALS so far was that these alterations emerge at some point in time shortly before overt symptomatic motor signs. The hereby presented data suggest a different concept for C9orf72-associated ALS. Both white matter alterations and verbal fluency in C9orf72 carriers might be indicative of general developmental deficits which are thus unrelated to time. Interestingly, developmental deficits early in life are a significant contributor to frailty and late-life health inequalities including dementia.37 In C9orf72 carriers, non-verbal memory was significantly impaired and there was evidence for a decline in memory performance (and attention) with age in our cohort. Thus, the impaired non-verbal memory performance might be an early sign in the sense of ageing-related changes.38
As detailed in the introduction, structural and functional changes on cortical level in association with C9orf72 may precede overt symptomatic onset.6–9 Other studies provided no evidence for cognitive changes in the preclinical state in presymptomatic C9orf72 expansion carriers despite overt signs of network degeneration9 or cognitive changes were associated with microstructural changes only in C9orf72 carriers younger than 40 years of age.39 When evaluating these earlier studies, it should be recalled that (a) the RPPCR (repeat-primed polymerase chain reaction) blood assay technique used in most laboratories to determine the presence of C9orf72HRE carries a significant proportion of both false-positive and false-negative cases15 and (b) autopsies of C9orf72HRE (ALS or FTD or both) have revealed extensive mosaicism with no correlation of expansion size (as determined by the gold standard method Southern blot) between blood and various parts of the brain.40 Hence, an individual with a significant expansion in blood leucocytes may necessarily not have an expansion in all parts of the brain or vice versa.
The underlying biological mechanism for this developmental deficit in C9orf72 carriers may be grounded in that the C9orf72 protein has been proposed to play an essential role in the development of the CNS11 and that C9orf72 carriers present with altered synaptic structures.12 Also, C9orf72 pathology is associated with cytoplasmic mislocalisation of TDP43 in the brain following predefined patterns.13 14 Further, it is most likely that complex disturbances of nuclear and cytoplasmic functions together with generation of toxic species may contribute to cell death in C9orf72 repeat expansion mutation even in the preclinical state (for review, see reference15).
Thus, the pattern in the executive function of verbal fluency associated with loss of structural integrity in C9orf72 carriers in orbitofrontal regions might be driven by pathological neuronal trafficking early in life, decades before any overt clinical signs of ALS (or FTD). Interestingly, orbitofrontal regions are the cortical regions first affected in FTD subjects,13 bringing C9orf72 carriers closer to structural FTD phenotypes.
Major strengths of the study are the large and well-characterised sample size and the inclusion of non-mutation carrier subjects from affected families. Both the study participants were blinded to their respective genotypes and also all evaluators of the neuropsychological tests and imaging were blinded. Given that FMs independent of their genetic status were more anxious, we have controlled our statistics for this confounder but the impact of anxiety on cognitive performance was negligible.
The current study has a number of limitations. The first of these is the inclusion of volunteers from families with a risk for ALS or FTD which raises the possibility of selection bias as volunteering participants generally tend to be of higher socio-economic status and better educated than non-participants.41 Further, subjects prone to FTD pathology might present with a preclinical manifestation of disinhibited behaviour which might increase the likelihood to participate in a study such as this. All these potential bias factors might limit the generalisability of the study results which thus need further replication in other populations. However, as no information on non-participants can be provided, any impact of selection bias can only be speculated on.
Overall, our results indicate that C9orf72 carriers present with cerebro-structural and neuropsychological dysfunctions which do not seem to be not solely associated with neurodegenerative processes at the time of clinical manifestation but might also involve developmental tardiness as a general trait of C9orf72 carriers. This latter aspect should be taken into consideration in future interventional trials in C9orf72-associated disorders.
Acknowledgments
We would like to thank all patients and carers who supported us by giving their valuable time for their participation in the study. We thank Sarah Böhm for study design and data collection, Jürgen Keller for support in data collection.
References
Footnotes
DEL and H-PM are joint first authors.
JK and ACL are joint senior authors.
Contributors DEL, PW, PA, JW: design or conceptualisation of the study, analysis or interpretation of the data, drafting or revising the manuscript for intellectual content. HPM, JF: analysis or interpretation of the data, drafting or revising the manuscript for intellectual content. AK: design or conceptualisation of the study. IW, JK, ACL: design or conceptualisation of the study, drafting or revising the manuscript for intellectual content. IU: drafting or revising the manuscript for intellectual content. A
Funding This is an EU Joint Programme—Neurodegenerative Disease Research (JPND) project ('NEEDSinALS' 01ED1405 and PreFrontAls 01ED1512). The project is supported through the following organisations under the aegis of JPND—www.jpnd.eu for example, Germany, Bundesministerium für Bildung und Forschung (BMBF, FKZ), Sweden, Vetenskaprådet Sverige, Poland, Narodowe Centrum Badań i Rozwoju (NCBR). This work was additionally funded by the Deutsche Forschungsgemeinschaft (DFG, LU 336/13–2), the Bundesministerium für Bildung und Forschung (FTLDc O1GI1007A, MND-Net 01GM1103A; PaCeMed 01DS18031) and the foundation of the state Baden-Württemberg (D.3830), Boehringer Ingelheim Ulm University BioCenter (D.5009), Thierry Latran Foundation. Also, this project is funded by the Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE), and the Knut and Alice Wallenberg Foundation.
Disclaimer The funding sources played no role in the preparation of this manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the Ethics Committee of the University of Ulm (reference 68/19), in accordance with the ethical standards of the current version of the revised Helsinki Declaration. All participants gave verbal and written informed consent prior to inclusion in the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Reasonable data sharing requests are made in writing through Dorothée Lulé (dorothee.lule@uni-ulm.de) and require a formal data sharing agreement. Data sharing agreements must include details on how the data will be stored, who will have access to the data and intended use of the data, and agreements as to the allocation of intellectual property.