Article Text

Abstract
Background There is limited evidence regarding optimal duration of antibiotic treatment in neuroborreliosis. We aimed to compare efficacy and safety of oral doxycycline for 2 and 6 weeks in European Lyme neuroborreliosis (LNB).
Methods The trial had a randomised, double-blinded, placebo-controlled, non-inferiority design. Patients with LNB were recruited from eight Norwegian hospitals and randomised to doxycycline 200 mg once daily for 2 weeks, followed by 4 weeks of placebo, or doxycycline 200 mg once daily for 6 weeks. The primary endpoint was clinical improvement as measured by difference in a Composite Clinical Score (0–64 points) from baseline to 6 months. The non-inferiority margin was predetermined to 0.5 points.
Results One hundred and twenty-one patients were included. Fifty-two treated for 2 weeks and 53 for 6 weeks were included in the intention-to-treat analyses, and 52 and 51 in per-protocol analysis. Mean difference in clinical improvement between the groups was 0.06, 95% CI −1.2 to 1.2, p=0.99 in the intention-to-treat population, and −0.4, 95% CI −1.4 to 0.7, p=0.51 in the per-protocol population and non-inferiority could not be established. There were no treatment failures and no serious adverse events. The groups did not differ in secondary outcomes including clinical scores at 10 weeks and 12 months, cerebrospinal fluid data and patient-reported outcome measures. Patients receiving 6 weeks doxycycline reported slightly more side effects in week 5.
Conclusion Our results strongly indicate that there are no benefits of doxycycline treatment beyond 2 weeks in European LNB.
Trial registration number 2015-001481-25.
- borrelia
- lyme disease
- infectious diseases
Data availability statement
Data are available on reasonable request. The data that support the findings of this study are available on request from the corresponding author with investigator support and after approval of a proposal to the BorrSci study group. The data are not publicly available due to privacy or ethical restrictions. The study protocol and informed consent form are freely available from the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
There is limited evidence for optimal treatment duration in Lyme neuroborreliosis (LNB), and clinical practice varies considerably. To the authors’ knowledge, there is only one previous randomised trial comparing treatment length in disseminated Lyme borreliosis (ie, symptoms that reflect that the infection has spread from the site of the tick bite to other parts of the body). Systematic reviews of LNB treatment call for more high-quality research on the matter.
WHAT THIS STUDY ADDS
This is the first randomised, placebo-controlled trial comparing treatment lengths of doxycycline on a well-defined patient population with European LNB.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Our findings support recommendations of 2 weeks doxycycline treatment in acute European LNB with typical manifestations.
Introduction
European Lyme neuroborreliosis (LNB) typically presents with painful meningoradiculitis and/or cranial neuritis, accompanied with malaise and fatigue. More rare clinical manifestations are plexus neuritis, mononeuritis and central nervous system (CNS) syndromes such as myelitis, vasculitis and encephalitis. It is an unambiguous agreement that patients with LNB should be treated with antibiotics as soon as possible, but both the choice of antibiotic type and treatment duration have been subjects for discussion.
Intravenously administered beta-lactam antibiotics (penicillin G, ceftriaxone and cefotaxime) and orally administered doxycycline are proven effective and hold relatively good cerebrospinal fluid (CSF) penetration.1 Orally administered doxycycline has been shown to be non-inferior to intravenous ceftriaxone in typical LNB,2 3 and probably effective in LNB with mainly CNS involvement.4 In line with this knowledge, European Federation of Neurological Societies (EFNS) guidelines from 20105 recommend treatment with either a beta-lactam antibiotic or doxycycline in adults. The final choice of antibiotic type depends on individual factors such as age, tolerability, pregnancy, breast-feeding and preferred mode of administration. According to the guidelines, patients with encephalitis, myelitis or vasculitis should be treated with intravenous ceftriaxone.
The recommendations regarding treatment duration are divergent. The EFNS guidelines recommend 2 weeks for early LNB, defined as pretreatment symptom duration under 6 months and 3 weeks for late LNB, defined as pretreatment symptom duration over 6 months.5 The German guidelines from 20206 are in accordance with EFNS guidelines, but the National Institute for Health and Care Excellence guidelines from 2018 recommend 3 weeks irrespective of symptom duration.7 These recommendations are mainly based on clinical experience, as scientific evidence is scarce. To our knowledge, there is only one previous randomised trial comparing treatment lengths in disseminated Lyme borreliosis. In that trial, including 62 with definite and 53 with possible LNB, the long-term outcome did not differ in patients treated for 3 weeks with intravenous ceftriaxone as compared with patients treated 3 weeks with intravenous ceftriaxone, followed by amoxicillin for 100 days.8 It is also noticeable that several studies have evaluated prolonged antibiotic treatment in patients suffering from so-called post-Lyme disease without demonstrating benefits.9–12
Irrespective of recommendations, there are clues to substantial variations in clinical practice. In a Norwegian study of adherence to guidelines, 61% of patients with LNB were treated for more than 2 weeks, 36% for more than 3 weeks and 12% for more than 6 weeks.13
Several factors are likely to influence choice of treatment duration, such as limited evidence in guidelines, local treatment cultures, beliefs and attitudes, and pressure and expectations from the patients due to advocacy in media for long duration of antibiotic treatment in LNB. High-quality research has been called for to pave the way for more evidence-based treatment decisions in the clinical practice.14 15
In light of all this, we aimed to compare efficacy and safety of treatment with oral doxycycline for 2 and 6 weeks in European LNB in a randomised controlled trial. We chose a non-inferiority approach for assessment of efficacy as the short antibiotic regimen was not expected to be superior to the long regimen, but it still offers clear advantages in terms of antibiotic resistance issues, lower costs and less strain on the patients.
Method
Study design
The trial has a randomised, double-blinded, placebo controlled, multicentre, non-inferiority design. For further details, we refer to the previously published study protocol.16 Adult patients were included from neurological or infectious diseases departments at eight hospitals in Southern Norway, with Sørlandet hospital in Agder county as coordinator.
Participants
We included consecutive patients with neurological symptoms suggestive of LNB without other obvious reasons and CSF pleocytosis and/or borrelia-specific antibodies produced intrathecally from hospital wards or outpatient clinics. In accordance with EFNS guidelines, the LNB was classified as possible in the presence of either CSF pleocytosis or borrelia-specific antibodies produced intrathecally, and as definite in the presence of both. All participants gave written informed consent before inclusion. Exclusion criteria are presented in the published protocol.16
Randomisation and masking
Patients were randomised into two treatment arms: oral doxycycline 200 mg once daily for 2 weeks, followed by 4 weeks of placebo, or doxycycline 200 mg once daily for 6 weeks. All patients received identically designed tablets and capsules for 6 weeks. Computerised allocation, with stratification according to hospital, was performed by an internet-based solution provided by the Department of Clinical Research Support, Oslo University Hospital. The Department of Clinical research also provided external monitoring of the procedures at all study centres according to good clinical practice. Patients, clinicians and study personnel were blinded to treatment allocation. The blinding was retained until all patients had completed the 6 months visit, the content of all tables and figures were fixed, and the statistical procedures were performed with the two treatment arms marked as groups A and B.
Outcomes
The study procedures are explained in the published protocol.16 Patients had outpatient follow-up at 10 weeks, 6 months and 12 months after inclusion, and additional blood samples were collected 2 and 4 weeks after the start of treatment. The patients were scored on a Composite Clinical Score (CCS) at each visit. The CSS measures 10 subjective symptoms and 22 objective neurological findings. Each of the 32 items is scored as 0=none, 1=mild (without influence on daily life) or 2=severe (with influence on daily life), and the sum score range from 0 to 64.16 Clinicians at each site scored patients, and discussed with study coordinators when necessary.
The primary endpoint was clinical improvement 6 months after treatment start as measured by difference in CCS sum score from baseline to 6 months.
Secondary endpoints were CCS at 10 weeks and 12 months, CSF findings at 6 and 12 months, safety and tolerability as measured by blood tests (haematological values, kidney and liver function) at 2 and 4 weeks after treatment start, patient-reported outcome measures (PROMs) and weekly reported side effects in a patient diary for 10 weeks. The PROMs were fatigue scored with Fatigue Severity Scale, subjective somatic symptoms scores with the Patient Health Questionnaire-15 and health-related quality of life (RAND 36). The patient diary consisted of five questions regarding side effects; nausea, diarrhoea, skin changes, genital ailments and decreased appetite. Each question was answered with 0=no symptoms, 1=mild symptoms or 2=severe symptoms, and a sum score was calculated each week ranging from 0 to 10.
Statistical analysis
The primary objective of the study was to determine if treatment duration of 2 weeks doxycycline is as effective as a prolonged regimen of 6 weeks. Accordingly, the null hypothesis was that 2 weeks treatment duration is inferior to 6 weeks treatment duration.
For the sample size calculation, the authors drafting the protocol considered a non-inferiority margin of 0.5 points in mean improvement on the CCS as clinically relevant. In other words, a mean difference in the clinical score from baseline to 6 months after inclusion of up to 0.5 points represented the maximum reduction in effectiveness we would accept, while still considering the short regimen treatment to be non-inferior. We used data from our previous trial with an adult population with European LNB and the same clinical score to calculate sample size and to determine the non-inferiority margin.2 With a significance level of 0.05 and statistical power at 80% this corresponded to a sample size of 50 patients in each group. To compensate for 20% drop-outs, we planned to include 120 patients, with 60 in each group.
For analyses of the primary endpoint, we applied a general linear model adjusting for gender, age, pretreatment duration of symptoms and CCS at baseline.
The primary endpoint was analysed in an intention-to-treat (ITT) principle population (excluding participants who withdrew consent, discontinued treatment and/or were lost to follow-up) and in a per-protocol population. In the latter, we also excluded one patient who fulfilled treatment according to protocol but was shown to have another diagnosis explaining the symptoms, and one patient who got a fatal additional disease and therefore scored unreasonably high on CCS.
Secondary endpoints, except safety issues, were compared between groups in the per-protocol population, applying independent samples t-tests, non-parametric tests or Pearson’s χ2 or Fisher’s exact test for cross tabs as appropriate with a two-sided CI approach. Values of p<0.05 were considered significant.
We used the statistical program SPSS V.26 for analysis.
Results
At least 144 consecutive patients with suspected LNB were assessed for eligibility in the study period from 20 November 2015 to 6 January 2020. Twenty-three were considered ineligible, 4 because clinicians outside the study already had initiated intravenous ceftriaxone (1 due to LNB cerebral vasculitis, 1 due to LNB with cognitive problems for longer than 6 months,and 2 for unknown reason), and 19 because they declined invitation, or met other exclusion criteria. As the screening log was complete only at Sørlandet hospital, it cannot be ruled out that additional patients were considered and found ineligible at the other centres.
A total of 121 patients were included and randomised to 2 or 6 weeks treatment with oral doxycycline, and 105 and 103 were included in statistical analysis according to ITT and per-protocol principle, respectively (figure 1). The 16 drop-outs were younger as compared with those included in the ITT analysis (mean age 48 vs 56 years, p=0.03), otherwise the baseline characteristic did not differ. Four of the six patients that withdrew consent were prescribed further antibiotics by their general practitioner (GP) after 2 weeks of treatment because of incomplete recovery. One additional patient received further antibiotics from the GP between the 10 weeks and 6 months follow-up. Study personnel evaluated all of these patients and found no new symptoms or findings consistent with treatment failure, and they did not differ in baseline CCS. Another patient developed suspected Lyme arthritis in a knee after 1 week of treatment and was prescribed additional 2 weeks of unblinded doxycycline. One patient had a much higher CCS score at 6 months follow-up as compared with baseline. Retrospectively, we consider this finding to be attributed to cancer disease, not LNB, as the patient at this point had been transferred to hospice care and died soon afterwards. This patient was excluded from the per-protocol analysis.
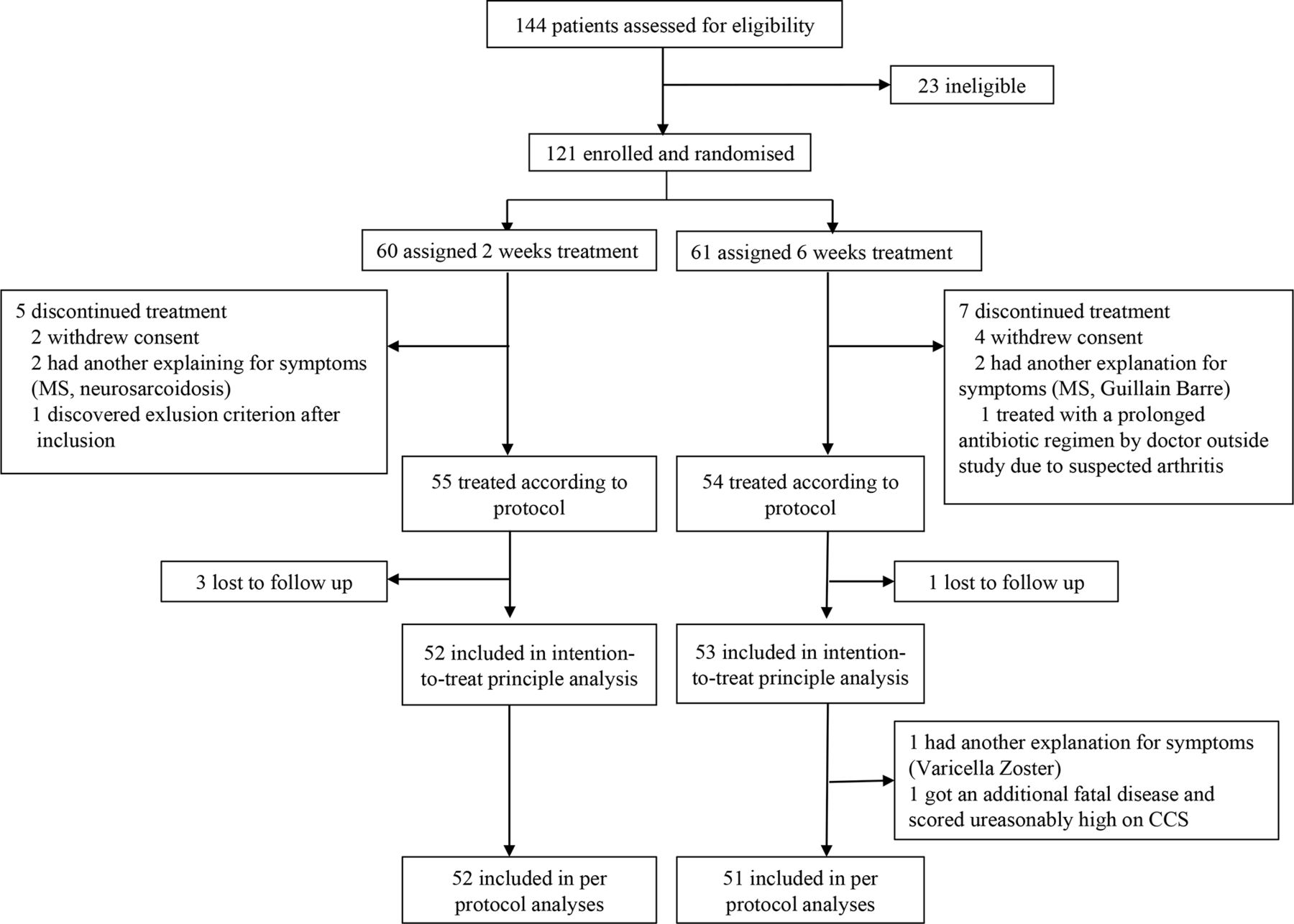
Flow chart of included patients. CCS, Composite Clinical Score. MS, Multiple Sclerosis
The baseline characteristics of the ITT population are shown in table 1. Among the 18 patients classified as possible LNB, 1 had normal CSF cell count but positive intrathecal antibody ratio, 2 had CSF pleocytosis but missing data on intrathecal antibody production and 15 had CSF pleocytosis, but negative intrathecal antibody ratio at inclusion and at 6 months after inclusion. Of these 15, 6 had negative borrelia antibodies also in serum (2 with only facial palsy in the 6 weeks treatment groups, 2 with facial palsy and other radiculitis in the 6 weeks treatment group, and 2 with radiculitis in the 2 weeks treatment group) throughout the study. Fifteen of the 18 were tested for tickborne encephalitis antibodies in serum with negative result, 15 were tested for herpes simplex and varicella zoster PCR in CSF of which 1 tested positive for varicella zoster (excluded from per-protocol analysis), and 12 were tested for enterovirus PCR in CSF with negative result.
Baseline demographics, clinical and laboratory findings of the intention to treat population. Data are number of patients (%) or mean (SD) unless otherwise stated
Only one patient had a confirmed LNB CNS syndrome with clinical and radiological signs of myelitis. Thirteen patients (seven in the 2 weeks treatment group and six in the 6 weeks treatment group) had scores that could indicate CNS involvement. The findings were subtle, however, mostly scored as mild, and none were confirmed by MRI: Mild central findings in one extremity (n=five), in hemipattern (n=one), or in both legs (n=one), mild dysphasia (n=one), mild cognitive impairment (n=three), and severe gait ataxia (n=one).
The primary endpoints in the two treatment groups are shown in table 2. The lower limit of the 95% CIs for difference between groups exceeded the predefined non-inferiority margin of 0.5 points, but the treatment groups were similar for superiority in both the ITT and per-protocol population. The two treatment groups were also similar with respect to clinical improvement in the subgroups definite and possible LNB (definite 6.3 vs 6.5, p=0.76 (mean difference=0.2, 95% CI −1.1 to 1.5) and possible 5.8 vs 5.9, p=0.95 (mean difference=0.08, 95% CI −2.9 to 2.7)) and with respect to all secondary endpoints (table 3 and figure 2).
Main outcome. clinical improvement 6 months after treatment start as measured by difference in clinical composite sum score from baseline to 6 months
Secondary endpoints

Composite Clinical Score throughout the study in the two treatment groups.
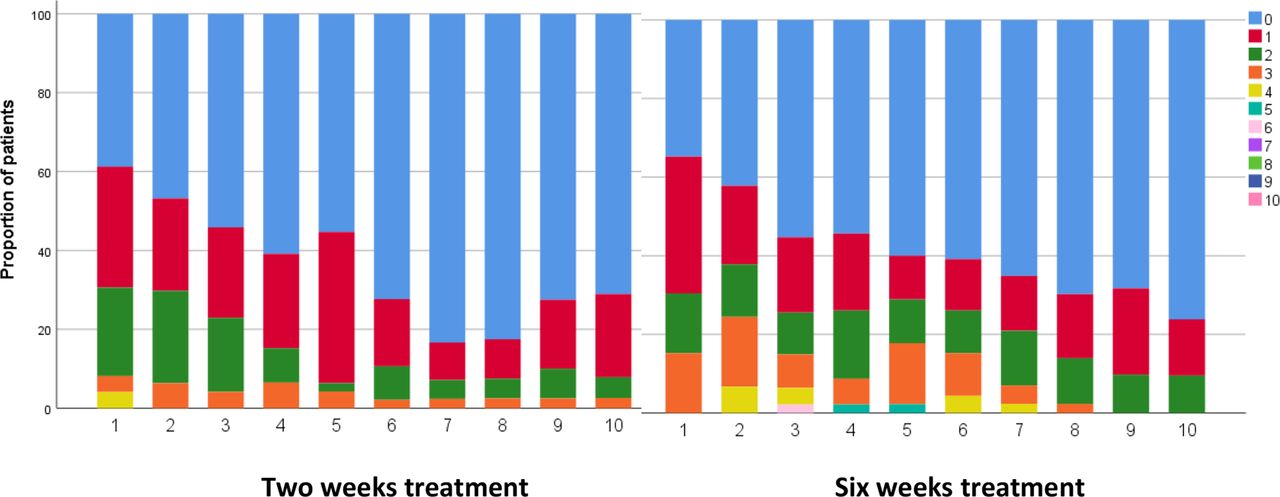
At 6 months, the proportions of patients with any complaint, including symptoms without influence on daily life was 73% and 71% in the 2 and 6 weeks treatment groups, respectively. The proportions with at least one complaint influencing daily life (CCS≥2) were 23% (n=12) and 22% (n=11). One patient treated for 6 weeks had an elevated CSF cell count at 6 months (baseline 82 M/L, 6 months 28 M/L), but had no other signs of treatment failure or disease progression, and the CSF cell count normalised at 12 months (3 M/L). We did not register any serious adverse events, and no patients were excluded, or had to stop treatment, because of adverse events. Weekly patient-reported side effects for 10 weeks are summarised in figure 3. There was a trend towards more patients with high sum scores in week 3–7 in the 6 weeks treatment group, but the difference was statistically significant only in week 5 (p=0.03). There were no difference in median sum scores between the treatment groups at any time point. The only single question that differed between the treatment groups was report of nausea at week 5, where nine patients reported mild and two serious nausea in the 6 weeks treatment group as compared with three patients with mild nausea in the 2 weeks treatment group (p=0.03). At week 10, two patients, one in each group, reported seriously decreased appetite, otherwise, the patients reported few and only mild remaining possible side effects. There were no statistically significant differences in frequencies of pathological blood sample findings between the two groups at 2 and 4 weeks after start of treatment.
{kind=link}
{kind=link}
{kind=link}
Side effects reported in patient diaries for ten weeks (week number on X axis), shown as proportion of patients each week with sum scores from 0 to 10 (none had sum score above 6) on five predefined symptoms (nausea, diarrhoea, skin changes, genital ailments and decreased appetite scored as 0 (none), 1 (mild), or 2 (severe)).
Discussion
In this randomised double-blinded treatment trial in European LNB, the primary outcome measure, improvement in a CSS at 6 months, did not differ between patients treated with 2 and 6 weeks of doxycycline neither when analysed in an ITT principle population, in a per-protocol population, nor in subgroups of patients with definite and possible LNB. Still, the lower bound of the 95% CI of the mean difference exceeded the predetermined non-inferiority margin of 0.5 points in both the ITT population and the per-protocol population, and we can, therefore, not claim statistical non-inferiority. Our results still strongly indicate that 6 weeks of doxycycline does not offer any benefits over 2 weeks in European LNB in adults.
This conclusion is supported by important findings beside the primary outcome. First, the treatment groups did not differ in any secondary outcomes including clinical scores at 10 weeks and 12 months, CSF data, and patient-reported outcomes on fatigue, subjective somatic symptoms scores and health-related quality of life. Second, we did not register any treatment failures in any of the two groups. The vast majority of patients improved well, but some had residual complaints. A proportion of 77% with any kind of residual complaint, and 17% with at least one residual complaint that influenced daily life at 12 months after start of treatment, is in accordance with earlier findings. Depending on scoring and sub-grouping, previous studies have found residual complaints in 24% to 48% at 12 months after treatment.17–20 As such, we do not regard the residual complaints as an indication of treatment failure, rather as sequelae in a few patients that is often found in this patient population, and we plan to evaluate possible prognostic markers in our cohort in future publications.
Regarding safety, we did not register any serious adverse events related to the treatment, and weekly median total score on self-reported side effects for ten weeks after start of treatment did not differ between the groups. In week five a higher proportion of patients receiving the longer course reported higher side effect sum scores and higher burden of nausea than patients receiving placebo, but our findings indicate that treatment with doxycycline is safe and rather well tolerated in prolonged treatment. In terms of antibiotic treatment, however, there is a consensus that “shorter is better” to decrease the burden of possible adverse effects, superinfections and microbial resistance.
In terms of trial limitations, there is a potential selection bias towards inclusion of patients with a less severe course of LNB, as some patients were found ineligible due to clinician or patient preferred choice of intravenous antibiotics. Such a choice could indicate that these patients had a higher symptom burden, longer pre-treatment symptom duration or confirmed CNS syndromes. At Sørlandet hospital, which had a complete and accurate screening log, and where 84 patients were included, only four patients were excluded due to choice of intravenous antibiotics. The proportion of patients excluded from trial participation due to a severe disease course may have been higher at the other study centres, but the screenings logs are unfortunately unreliable. In the final analyses, three patients had pretreatment symptom duration over 6 months, 44 had clinical score >10, 12 had subtle findings indicating possible CNS involvement, and one had a confirmed CNS syndrome (myelitis). In light of this, we think a possible selection bias regarding overall symptom burden is ignorable, but possible regarding patients with long pre-treatment symptom duration or LNB with manifestations such as myelitis, vasculitis or encephalitis. Patients with late LNB or with confirmed CNS syndromes are very rare however, and a randomised controlled trial in these subgroups would be very resource-intensive and time consuming. Regardless, cautions should be made in treatment recommendations for patients with late LNB and patients with confirmed CNS syndromes.
Another possible limitation is the use of an unvalidated clinical score (CCS) that carries both inter-and intra-observer variability. The randomisation procedure is assumed to equalise this variability, but still the score is encumbered with imprecise absolute scores that may cause wide 95% CIs. We chose the score since we were familiar with it from a previous trial,2 it reflects assessments done of these patients in clinical practice, and it has also been used in a modified form by other researchers.19
Our study included 18 patients with possible LNB including six with negative antibodies in both serum and CSF. A relatively short pretreatment symptom duration, ranging from two to 7 days, could explain persistent antibody negativity in these six patients,21 22 but it is possible that some of them did not have LNB. Two had facial palsy as their only symptom, and retrospectively thought to have suffered from Bell’s palsy. The inclusion of some patients with unclear diagnosis is unavoidable in a treatment trial of LNB due to low sensitivity of intrathecal antibody production in the early phase of the disease, and the need to start antibiotic treatment before antibody results are available. This study included relatively few such patients, however.
Overall, we consider the trial to be well designed with few sources of biases and high internal validity. We also think the trial results reflect everyday clinical practice by including patients with both definite and possible LNB from different centres, and thereby possess high external validity.
Conclusion
We could not establish statistical non-inferiority using the pre-specified margins when comparing efficacy on clinical improvement after 6 months in 2 and 6 weeks treatment with oral doxycycline in European LNB. Still, our study results strongly indicate that there is no added benefit of treatment beyond the 2 weeks in current guidelines.
Supplemental material
Data availability statement
Data are available on reasonable request. The data that support the findings of this study are available on request from the corresponding author with investigator support and after approval of a proposal to the BorrSci study group. The data are not publicly available due to privacy or ethical restrictions. The study protocol and informed consent form are freely available from the corresponding author.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Norwegian Regional Committees for Medical and Health Research Ethics, 2015/1031. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank all our collaborators at each of the eight hospitals for facilitating the study, recruiting patients and collecting data: Anna Wie Børsheim at Haukeland University Hospital, Marion Jim at Vestre Viken Hospital, Marie Mørck Møinichen at Vestfold Hospital, Pascal Brügger-Synnes at Møre and Romsdal Hospital trust. We also thank Hanne Quarsten, Sølvi Noraas and Nils Arne Tryland at the departments of microbiology and biochemistry at Sørlandet Hospital for supporting our work and handling collected samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ÅM and UL are joint senior authors.
ÅM and UL contributed equally.
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors AMS collected data from the primary inclusion site and facilitated at the additional sites, wrote revisions on the study protocol, did the statistical analysis, wrote the first drafts of the manuscript and acted as guarantor with input from UL and ÅM. ÅRL collected data from the primary inclusion site, facilitated bio banking of study material, and contributed to the reviewing and editing of the manuscript. AOD collected data from inclusion site, contributed to the reviewing and editing of the manuscript. HØF collected data from inclusion site, contributed to the reviewing and editing of the manuscript. SB collected data from inclusion site, contributed to the reviewing and editing of the manuscript. KJNF collected data from the primary inclusion site, and contributed to the reviewing of the manuscript. AHP contributed to data analysis and statistical analysis. MHB collected data from inclusion site, and contributed to the reviewing of the manuscript. RE contributed to the study protocol, was active in the study inclusion and organisation process, data analysis and contributed to the reviewing of the manuscript. HR contributed to the conceptualisation, project administration, resources, validation, visualisation and the reviewing and editing of the manuscript. ÅM designed the trial, wrote the first draft of the protocol, acquired funding, collected data from the primary inclusion site, contributed to the statistical analyses and writing of the first drafts of the manuscript. UL designed the trial, wrote the first draft of the protocol, acquired funding, collected data from the primary inclusion site, did the statistical analyses and contributed to the writing of the first draft of the manuscript. UL and ÅM contributed equally to the trial and manuscript and share senior authorship.
Funding The Norwegian Multiregional Health Authorities funded the study through the Borrsci project (Lyme borreliosis; a scientific approach to reduce diagnostic and therapeutic uncertainties, project 2015113). ARL received funding by the South-Eastern Norway Regional Health Authority (project 2013089).
Competing interests SB has received honoraria for lecturing from Biogen and Novartis.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.