Article Text
Research Article
MRI of neuromyelitis optica: evidence for a distinct entity.

Statistics from Altmetric.com
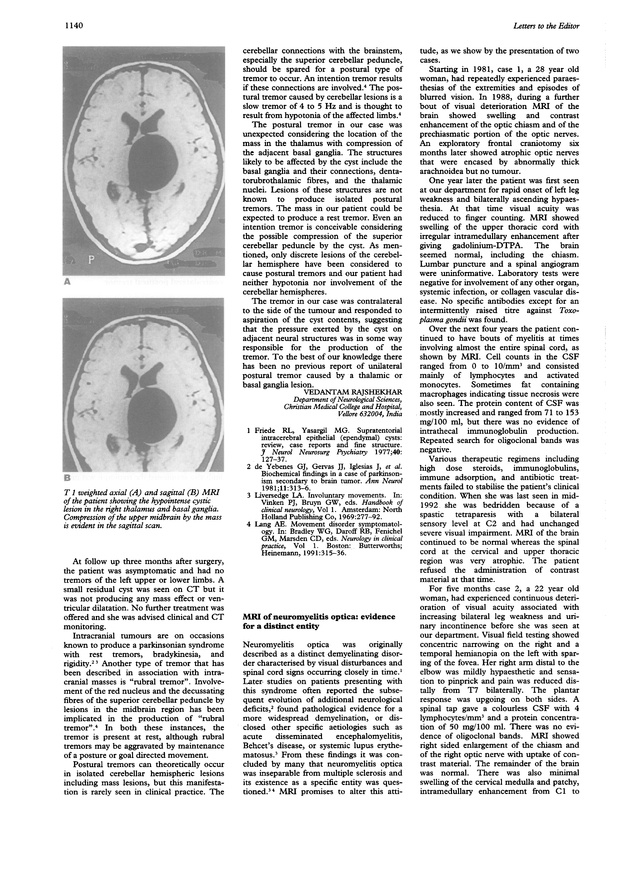
This is a PDF-only article. The first page of the PDF of this article appears above.
![]()
Article Text
This is a PDF-only article. The first page of the PDF of this article appears above.