
Abstract
Purpose
To test in a longitudinal follow-up study whether basal glucose metabolism in subjects with a genetic risk of Huntington disease (HD) may influence the onset of manifest symptoms.
Methods
The study group comprised 43 presymptomatic (preHD) subjects carrying the HD mutation. They underwent a 18F-FDG PET scan and were prospectively followed-up for at least 5 years using the unified HD rating scale to detect clinical changes. Multiple regression analysis included subject’s age, CAG mutation size and glucose uptake as variables in a model to predict age at onset.
Results
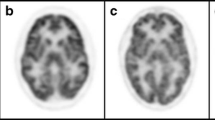
Of the 43 preHD subjects who manifested motor symptoms, suggestive of HD, after 5 years from the PET scan, 26 showed a mean brain glucose uptake below the cut-off of 1.0493 in the caudate, significantly lower than the 17 preHD subjects who remained symptom-free (P < 0.0001). This difference was independent of mutation size. Measurement of brain glucose uptake improved the CAG repeat number and age-based model for predicting age at onset by 37 %.
Conclusion
A reduced level of glucose metabolism in the brain caudate may represent a predisposing factor that contributes to the age at onset of HD in preHD subjects, in addition to the mutation size.


Similar content being viewed by others


References
Grafton ST, Mazziotta JC, Pahl JJ, St George-Hyslop P, Haines JL, Gusella J, et al. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch Neurol. 1992;49:1161–7.
Kuwert T, Lange HW, Boecker H, Titz H, Herzog H, Aulich A, et al. Striatal glucose consumption in chorea-free subjects at risk of Huntington's disease. J Neurol. 1993;241:31–6.
Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain. 1996;119(Pt 6):2085–95.
Feigin A, Fukuda M, Dhawan V, Przedborski S, Jackson-Lewis V, Mentis MJ, et al. Metabolic correlates of levodopa response in Parkinson's disease. Neurology. 2001;57:2083–8.
Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med. 2006;47:215–22.
Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60:1202–10.
Squitieri F, Sabbadini G, Mandich P, Gellera C, Di Maria E, Bellone E, et al. Family and molecular data for a fine analysis of age at onset in Huntington disease. Am J Med Genet. 2000;95:366–73.
Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. 2004;65:267–77. doi:10.1111/j.1399-0004.2004.00241.x.
Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet. 1996;59:16–22.
Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101:3498–503. doi:10.1073/pnas.0308679101.
Esmaeilzadeh M, Ciarmiello A, Squitieri F. Seeking brain biomarkers for preventive therapy in Huntington disease. CNS Neurosci Ther. 2011;17:368–86. doi:10.1111/j.1755-5949.2010.00157.x.
Penney Jr JB, Young AB, Shoulson I, Starosta-Rubenstein S, Snodgrass SR, Sanchez-Ramos J, et al. Huntington's disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals. Mov Disord. 1990;5:93–9.
Lynoe N, Sandlund M, Dahlqvist G, Jacobsson L. Informed consent: study of quality of information given to participants in a clinical trial. BMJ. 1991;303:610–3.
Huntington Study Group. Unified Huntington's disease rating scale: reliability and consistency. Mov Disord. 1996;11:136–42.
Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126:946–55.
Alfano B, Brunetti A, Covelli EM, Quarantelli M, Panico MR, Ciarmiello A, et al. Unsupervised, automated segmentation of the normal brain using a multispectral relaxometric magnetic resonance approach. Magn Reson Med. 1997;37:84–93.
Quarantelli M, Berkouk K, Prinster A, Landeau B, Svarer C, Balkay L, et al. Integrated software for the analysis of brain PET/SPECT studies with partial-volume-effect correction. J Nucl Med. 2004;45:192–201.
Muller-Gartner HW, Links JM, Prince JL, Bryan RN, McVeigh E, Leal JP, et al. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. J Cereb Blood Flow Metab. 1992;12:571–83. doi:10.1038/jcbfm.1992.81.
Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39:904–11.
Minoshima S, Frey KA, Foster NL, Kuhl DE. Preserved pontine glucose metabolism in Alzheimer disease: a reference region for functional brain image (PET) analysis. J Comput Assist Tomogr. 1995;19:541–7.
Brickman AM, Buchsbaum MS, Shihabuddin L, Hazlett EA, Borod JC, Mohs RC. Striatal size, glucose metabolic rate, and verbal learning in normal aging. Brain Res Cogn Brain Res. 2003;17:106–16.
Petit-Taboue MC, Landeau B, Desson JF, Desgranges B, Baron JC. Effects of healthy aging on the regional cerebral metabolic rate of glucose assessed with statistical parametric mapping. Neuroimage. 1998;7:176–84. doi:10.1006/nimg.1997.0318.
Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36.
Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. doi:10.1016/S1474-4422(09)70170-X.
Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi:10.1016/S0092-8674(00)80623-6.
Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63:66–72.
Ruocco HH, Bonilha L, Li LM, Lopes-Cendes I, Cendes F. Longitudinal analysis of regional grey matter loss in Huntington disease: effects of the length of the expanded CAG repeat. J Neurol Neurosurg Psychiatry. 2008;79:130–5. doi:10.1136/jnnp.2007.116244.
Squitieri F, Cannella M, Simonelli M, Sassone J, Martino T, Venditti E, et al. Distinct brain volume changes correlating with clinical stage, disease progression rate, mutation size, and age at onset prediction as early biomarkers of brain atrophy in Huntington's disease. CNS Neurosci Ther. 2009;15:1–11. doi:10.1111/j.1755-5949.2008.00068.x.
Powers WJ, Videen TO, Markham J, McGee-Minnich L, Antenor-Dorsey JV, Hershey T, et al. Selective defect of in vivo glycolysis in early Huntington's disease striatum. Proc Natl Acad Sci U S A. 2007;104(8):2945–9. doi:10.1073/pnas.0609833104.
Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–12. doi:10.1083/jcb.200508072.
Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–81.
Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–38. doi:10.1016/j.cell.2004.06.018.
Battaglia G, Cannella M, Riozzi B, Orobello S, Maat-Schieman ML, Aronica E, et al. Early defect of transforming growth factor beta1 formation in Huntington's disease. J Cell Mol Med. 2011;15:555–71. doi:10.1111/j.1582-4934.2010.01011.x.
Gonitel R, Moffitt H, Sathasivam K, Woodman B, Detloff PJ, Faull RL, et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci U S A. 2008;105:3467–72. doi:10.1073/pnas.0800048105.
Cannella M, Maglione V, Martino T, Ragona G, Frati L, Li GM, et al. DNA instability in replicating Huntington's disease lymphoblasts. BMC Med Genet. 2009;10:11. doi:10.1186/1471-2350-10-11.
Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, et al. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18:3039–47. doi:10.1093/hmg/ddp242.
Li JL, Hayden MR, Almqvist EW, Brinkman RR, Durr A, Dode C, et al. A genome scan for modifiers of age at onset in Huntington disease: the HD MAPS study. Am J Hum Genet. 2003;73:682–7. doi:10.1086/378133.
Acknowledgments
We are grateful for the support of the Italian association of HD families 'Associazione-Italiana-Corea-di-Huntington-Neuromed' (funds from 5x1000, to F.S.), IRCCS Neuromed (institutional funds from 5x1000 to F.S. and S.O.), Ministry of Health, Italy (Ricerca Corrente, to F.S.), Italian Olympic Committee (CONI, to E.F.), and of the European Huntington’s Disease Network for the REGISTRY Study (to F.S., E.F. and S.O.).
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ciarmiello, A., Giovacchini, G., Orobello, S. et al. 18F-FDG PET uptake in the pre-Huntington disease caudate affects the time-to-onset independently of CAG expansion size. Eur J Nucl Med Mol Imaging 39, 1030–1036 (2012). https://doi.org/10.1007/s00259-012-2114-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-012-2114-z