
Abstract
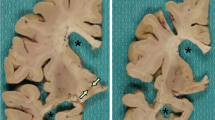
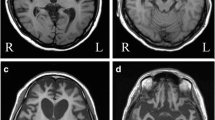
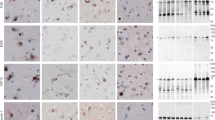
Basophilic Inclusion Body Disease (BIBD) is a tau-negative form of frontotemporal lobar degeneration (FTLD), characterized by neuronal cytoplasmic inclusions (NCI) that are visible on hematoxylin and eosin stain (HE), contain RNA, and are inconsistently ubiquitin-immunoreactive (ir). The normal nuclear expression of TDP-43 is not altered. Here we investigate whether the distribution of the structurally and functionally related protein fused in sarcoma (FUS) is altered in BIBD. Mutations in the FUS gene have recently been identified as a cause of familial amyotrophic lateral sclerosis (ALS). In addition to these familial ALS cases, FUS protein has recently been demonstrated in NCI in a subset of FTLD with ubiquitinated inclusions (atypical FTLD-U) and in neuronal intermediate filament inclusion disease (NIFID). We examined seven BIBD brains of patients with average age at onset 46 (range 29–57) and average duration of disease 8 years (range 5–12). Three cases presented with the behavioural variant of fronto-temporal dementia (FTD-bv) and one with FTD-bv combined with severe dysarthria. All four developed motor neuron disease/ALS syndrome (MND/ALS) several years later. In the other three cases, presentation was predominantly with motor symptoms, construed as MND/ALS in two, and progressive supranuclear palsy (PSP) in one. Severity of cortical degeneration varied, but all cases shared severe nigrostriatal atrophy and lower motor neuron pathology. In spared areas of cortex, FUS antibodies showed intense labelling of neuronal nuclei and weak positivity of cytoplasm, whereas, in affected areas, intense labelling of NCI was accompanied by reduction or disappearance of the normal IR pattern. The number of FUS-ir NCI was much greater than the number detected by HE or with ubiquitin or P62 immunohistochemistry. FUS-ir glial cytoplasmic inclusions (GCI) were abundant in the grey and white matter in all cases, whereas neuronal intranuclear inclusions were rare and only seen in 2/7 cases. Thus, BIBD shares with atypical FTLD-U and NIFID the presence of FUS-ir NCI and GCI, and together comprise a new biochemical category of neurodegenerative disease (FUS proteinopathies). The consistent involvement of motorneurons in BIBD indicates that the association of FTLD and MND/ALS can occur on a FUS or TDP-43 pathological substrate.







Similar content being viewed by others
References
Aizawa H, Kimura T, Hashimoto K et al (2000) Basophilic cytoplasmic inclusions in a case of sporadic juvenile amyotrophic lateral sclerosis. J Neurol Sci 176:109–113
Aman P, Panagopoulos I, Lassen C et al (1996) Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 37:1–8
Andersson MK, Stahlberg A, Arvidsson Y et al (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 9:37
Arai T, Hasegawa M, Akiyama H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Baechtold H, Kuroda M, Sok J et al (1999) Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem 274:34337–34342
Benajiba L, Le BI, Camuzat A et al (2009) TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65:470–473
Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS (1999) Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 18:4515–4521
Borroni B, Bonvicini C, Alberici A et al (2009) Mutation within TARDBP leads to Frontotemporal Dementia without motor neuron disease. Hum Mutat
Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8:776–789
Cairns NJ, Bigio EH, Mackenzie IR et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 114:5–22
Cairns NJ, Neumann M, Bigio EH et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Davies RR, Hodges JR, Kril JJ et al (2005) The pathological basis of semantic dementia. Brain 128:1984–1995
Fujii R, Okabe S, Urushido T et al (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15:587–593
Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118:5755–5765
Fujita K, Ito H, Nakano S et al (2008) Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol 116:439–445
Greenway MJ, Andersen PM, Russ C et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38:411–413
Gros-Louis F, Gaspar C, Rouleau GA (2006) Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:956–972
Hamada K, Fukazawa T, Yanagihara T et al (1995) Dementia with ALS features and diffuse Pick body-like inclusions (atypical Pick’s disease?). Clin Neuropathol 14:1–6
Hilton DA, McLean B (2002) December 2001: rapidly progressive motor weakness, starting in pregnancy. Brain Pathol 12:267–268
Hodges JR, Davies RR, Xuereb JH et al (2004) Clinicopathological correlates in frontotemporal dementia. Ann Neurol 56:399–406
Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 66:884–891
Ishihara K, Araki S, Ihori N et al (2006) An autopsy case of frontotemporal dementia with severe dysarthria and motor neuron disease showing numerous basophilic inclusions. Neuropathology 26:447–454
Jin P, Alisch RS, Warren ST (2004) RNA and microRNAs in fragile X mental retardation. Nat Cell Biol 6:1048–1053
Josephs KA, Lin WL, Ahmed Z et al (2008) Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116:159–167
Kertesz A, Blair M, McMonagle P, Munoz DG (2007) The diagnosis and course of frontotemporal dementia. Alzheimer Dis Assoc Disord 21:155–163
Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG (2005) The evolution and pathology of frontotemporal dementia. Brain 128:1996–2005
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665
Kusaka H, Matsumoto S, Imai T (1993) Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuropathol 12:215–218
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Leigh PN, Whitwell H, Garofalo O et al (1991) Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 114:775–788
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079
Mackenzie IR, Bigio EH, Ince PG et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Mackenzie IR, Foti D, Woulfe J, Hurwitz TA (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293
Mackenzie IR, Neumann M, Bigio EH et al (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18
Mackenzie IR, Rademakers R (2008) The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 21:693–700
Matsumoto S, Kusaka H, Murakami N et al (1992) Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol (Berl) 83:579–583
McKhann GM, Albert MS, Grossman M et al (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58:1803–1809
Mizutani T, Sakamaki S, Tsuchiya N et al (1992) Amyotrophic lateral sclerosis with ophthalmoplegia and multisystem degeneration in patients on long-term use of respirators. Acta Neuropathol 84:372–377
Munoz DG (1998) The pathology of Pick complex. In: Kertesz A, Munoz DG (eds) Pick’s disease and Pick complex. Wiley-Liss, New York, pp 211–241
Munoz-Garcia D, Ludwin SK (1984) Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 16:467–480
Murayama S, Mori H, Ihara Y et al (1990) Immunocytochemical and ultrastructural studies of lower motor neurons in amyotrophic lateral sclerosis. Ann Neurol 27:137–148
Neary D, Snowden JS, Gustafson L et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Nelson JS, Prensky AL (1972) Sporadic juvenile amyotrophic lateral sclerosis. A clinicopathological study of a case with neuronal cytoplasmic inclusions containing RNA. Arch Neurol 27:300–306
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain (in press)
Neumann M, Roeber S, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol (Berl). doi:10.1007/s00401-009-0581-5
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Oda M, Akagawa N, Tabuchi Y, Tanabe H (1978) A sporadic juvenile case of the amyotrophic lateral sclerosis with neuronal intracytoplasmic inclusions. Acta Neuropathol (Berl) 44:211–216
Okamoto K, Murakami N, Kusaka H et al (1992) Ubiquitin-positive intraneuronal inclusions in the extramotor cortices of presenile dementia patients with motor neuron disease. J Neurol 239:426–430
Padovani A, Agosti C, Premi E, Bellelli G, Borroni B (2007) Extrapyramidal symptoms in Frontotemporal Dementia: prevalence and clinical correlations. Neurosci Lett 422:39–42
Pikkarainen M, Hartikainen P, Alafuzoff I (2008) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol 67:280–298
Prasad DD, Ouchida M, Lee L, Rao VN, Reddy ES (1994) TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t(16;21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene 9:3717–3729
Rademakers R, Hutton M (2007) The genetics of frontotemporal lobar degeneration. Curr Neurol Neurosci Rep 7:434–442
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Sam M, Gutmann L, Schochet SS Jr, Doshi H (1991) Pick’s disease: a case clinically resembling amyotrophic lateral sclerosis. Neurology 41:1831–1833
Sasaki S, Toi S, Shirata A et al (2001) Immunohistochemical and ultrastructural study of basophilic inclusions in adult-onset motor neuron disease. Acta Neuropathol 102:200–206
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Takeda T, Uchihara T, Arai N, Mizutani T, Iwata M (2009) Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: distinction from Alzheimer disease. Acta Neuropathol 117:35–44
Tsuchiya K, Ishizu H, Nakano I et al (2001) Distribution of basal ganglia lesions in generalized variant of Pick’s disease: a clinicopathological study of four autopsy cases. Acta Neuropathol (Berl) 102:441–448
Tsuchiya K, Matsunaga T, Aoki M et al (2001) Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol 20:53–59
Valdmanis PN, Daoud H, Dion PA, Rouleau GA (2009) Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep 9:198–205
Vance C, Rogelj B, Hortobagyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Wang IF, Wu LS, Chang HY, Shen CK (2008) TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 105:797–806
Wilson CM, Grace GM, Munoz DG, He BP, Strong MJ (2001) Cognitive impairment in sporadic ALS: a pathologic continuum underlying a multisystem disorder. Neurology 57:651–657
Yang L, Embree LJ, Tsai S, Hickstein DD (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273:27761–27764
Yokota O, Tsuchiya K, Terada S et al (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115:561–575
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110(Pt 15):1741–1750
Acknowledgments
This work was supported by grants from Canadian Institutes of Health Research (DM, IM); the Pacific Alzheimer Research Foundation (IM); the Deutsche Forschungsgemeinschaft (MN); the Stavros-Niarchos Foundation (MN); and the Synapsis Foundation (MN). We thank Margaret Luk, Mareike Schroff, Nahid Nelson and Vidya Beharry for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Munoz, D.G., Neumann, M., Kusaka, H. et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118, 617–627 (2009). https://doi.org/10.1007/s00401-009-0598-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0598-9