




Abstract
Lesions of Baló's concentric sclerosis are characterized by alternating layers of myelinated and demyelinated tissue. The reason for concentric demyelination in this variant of multiple sclerosis is unclear. In the present study we investigated the immunopathology in autopsy tissue of 14 patients with acute multiple sclerosis or fulminant exacerbations of chronic multiple sclerosis with Baló-type lesions in the CNS, focusing on the patterns of tissue injury in actively demyelinating lesions. We found that all active concentric lesions followed a pattern of demyelination that bears resemblances to hypoxia-like tissue injury. This was associated with high expression of inducible nitric oxide synthase in macrophages and microglia. At the edge of active lesions and, less consistently, in the outermost layer of preserved myelin, proteins involved in tissue preconditioning, such as hypoxia-inducible factor 1α and heat-shock protein 70, were expressed mainly in oligodendrocytes and to a lesser degree also in astrocytes and macrophages. Due to their neuroprotective effects, the rim of periplaque tissue, where these proteins are expressed, may be resistant to further damage in an expanding lesion and may therefore remain as a layer of preserved myelinated tissue.
Introduction
Baló's concentric sclerosis (BCS) is considered a variant of inflammatory demyelinating disease closely related to multiple sclerosis. Its pathological hallmarks are large demyelinated lesions characterized by a peculiar pattern of alternating layers of preserved and destroyed myelin. Clinically, BCS is most often characterized by an acute onset with steady progression to major disability and death within months, thus resembling Marburg's acute multiple sclerosis (Marburg, 1906; Courville, 1970; Kuroiwa, 1985). However, more benign disease courses with only mild residual deficits have been reported (Revel et al., 1993; Korte et al., 1994; Ng et al., 1999) and smaller areas of alternate demyelination can be found in lesions of multiple sclerosis patients, who otherwise present with a more classical acute or chronic disease course (Marburg, 1906). T2-weighted MRI scans sometimes reveal a distinct pattern of hypo-/isointense and hyperintense rings corresponding to bands of preserved and destroyed myelin (Garbern et al., 1986; Spiegel et al., 1989; Miyata et al., 1990; Hanemann et al., 1993; Gharagozloo et al., 1994; Korte et al., 1994; Chen et al., 1999; Karaarslan et al., 2001). The characteristic pathological features were first described by Otto Marburg (Marburg, 1906) in a case of fulminant multiple sclerosis and later by Joseph Baló (Baló, 1927), reporting a patient with large hemispherical lesions entirely consisting of alternating layers of preserved and destroyed myelin (Marburg, 1906; Baló, 1927, 1928).
Various theories have been brought forward to explain this conspicuous pattern of demyelination. Colloid physicochemical processes favouring the precipitation of some unknown toxin or lecithinolytic factor have been proposed, resembling the precipitation of salts in a gel (the so-called Liesegang rings) (Hallervorden and Spatz, 1933). Ferraro demonstrated that the injection of potassium cyanide in rabbits produced pathological features resembling BCS (Ferraro, 1933). Courville stressed the resemblance of BCS lesions to patterns observed at the edge of hypoxic brain lesions, suggesting a vascular basis for the alternating layers present in BCS (Courville, 1970). Moore and colleagues proposed that BCS closely resembled acute multiple sclerosis except for an astounding intrinsic capacity of rapid remyelination, leading to the bands of myelin being separated by still fully demyelinated tissue (Moore et al., 1985). However, in another detailed study on concentric lesions, evidence for remyelination was lacking, and instead emphasis was laid on oligodendrocyte pathology (Yao et al., 1994). There, the authors found myelin-forming cells with pycnotic nuclei suggestive of apoptosis and hypothesized that the main pathogenetic insult might be directed against the oligodendrocyte.
Recent immunopathological studies of multiple sclerosis tissue suggest that the pathogenetic mechanisms leading to the formation of the demyelinated plaque may be heterogeneous. Multiple sclerosis lesions with T-cell/macrophage infiltration alone, with antibody and complement deposition, and with primary oligodendrocyte damage, some of them presenting with features suggestive of hypoxia-like tissue injury, have been identified (Lucchinetti et al., 2000; Aboul-Enein et al., 2003). In this study, we examined 14 cases of acute or fulminant chronic multiple sclerosis with lesional pathologies consistent with BCS. Whenever active demyelination occurred in these lesions, characteristic pathological features were present. The lesions were found to expand radially and to be centred on an inactive lesional core. The pattern of active demyelination and tissue injury closely resembled that found in acute hypoxic/ischaemic white matter injury, namely oligodendrocyte apoptosis and loss of myelin associated glycoprotein (MAG) (Aboul-Enein et al., 2003). In addition, proteins involved in tissue preconditioning were upregulated at the edge of actively demyelinating lesions and in part in the outer myelinated layers of the concentric lesion. These results suggest that preconditioned oligodendrocytes may be resistant to further damage at the edge of radially expanding lesions, leading to the concentric preservation of myelin found in BCS.
Material and methods
Cases and neuropathological classification
Brain tissue from 14 (nine female, five male) cases of multiple sclerosis with concentric lesions were included in this study, comprising 10 cases with acute multiple sclerosis and four chronic multiple sclerosis cases with a fulminant terminal exacerbation. The cases were derived from different geographical areas (central Europe, seven; USA and Canada, three; the Philippines, four). The age of the acute multiple sclerosis patients ranged from 17 to 45 years and the disease duration from 1 week to 6 months. The four patients with chronic multiple sclerosis (three females, one male; age 22–46 years; 3–5 years disease duration) had a fulminant terminal attack of the disease with a duration of the current illness of 2–4 weeks.
Patients with concentric lesions were compared with six patients with acute multiple sclerosis, 12 patients with chronic active multiple sclerosis and 10 patients with chronic inactive multiple sclerosis, who lacked concentric demyelination. In addition, we included brain autopsies from six patients with acute stroke lesions in the white matter. The demographic data of these patients have been published previously (Aboul-Enein et al., 2003).
The tissue was fixed in buffered formalin and routinely embedded in paraffin. Sections stained with haematoxylin–eosin, Luxol fast blue/periodic acid Schiff reaction (LFB/PAS) and Bielschowsky silver impregnation were used for standard neuropathological evaluation. Demyelinating activity within the lesions was determined by the presence of macrophages containing early myelin degradation products according to Brück and colleagues (Brück et al., 1995). The neuropathological studies were carried out according to the national ethical guidelines and legal regulations regarding the use of archival post-mortem material.
Immunohistochemistry
The antibodies used for immunohistochemistry in this study were directed against different myelin proteins, oligodendrocytes, astrocytes, microglia and macrophages, T-cells, B-cells, hypoxia-related proteins (HIF-1α, D-110, hsp70) and inducible nitric oxide synthase (iNOS) (Table 1). Primary antibodies were detected using biotinylated secondary antisera followed by avidin–peroxidase (Vass et al., 1986). Diaminobenzidine was used as the chromogenic substrate. Alternatively, we used a system based on alkaline phosphatase–anti-alkaline-phosphatase, with Fast Red for visualization of bound primary antibody (Vass et al., 1988). Quantitative analysis of immunostained cells was performed on consecutive serial sections. The number of cells was determined in zones of active myelin destruction in 10 standardized microscopic fields of 10 000 µm2, and the values were recalculated as cells per mm2 of tissue.
Antibodies used for immunocytochemistry
| Antibody | Clone | Dilution | Pretreatment | Source |
|---|---|---|---|---|
| PLP | Rabbit pk | 1 : 1000 | MW | Serotec, Oxford, UK |
| MBP | Mouse IgG1 | 1 : 500 | MW | Boehringer-Mannheim, Mannheim, Germany |
| MAG | D7E10 | 1 : 4000 | MW | Dobersen et al., 1985 |
| MOG | 8–18C5 | 1 : 500 | MW | Linington et al., 1988 |
| CNPase | SMI91 | 1 : 2000 | MW | Sternberger Monoclonals, MD, USA |
| D-110 | D-110 | 1 : 5000 | MW | Lassmann et al., 2003 |
| HIF-1α | Clone 54 | 1 : 30 | MW | BD Transduction Laboratories, Lexington, KY, USA |
| hsp70 | C92F3A-5 | 1 : 200 | MW | StressGen, Victoria, Canada |
| CD3 | CD3-12 | 1 : 200 | MW | Serotec |
| CD20 | L 26 | 1 : 50 | MW | Dako, Glostrup, Denmark |
| CD68 | KP1 | 1 : 100 | MW | Dako, Glostrup, Denmark |
| iNOS | AB 5384 | 1 : 30.000 | MW | Chemicon, Temecula, USA |
| Caspase-3 | C92-603 | 1 : 5000 | MW | BD Biosciences, USA |
| Antibody | Clone | Dilution | Pretreatment | Source |
|---|---|---|---|---|
| PLP | Rabbit pk | 1 : 1000 | MW | Serotec, Oxford, UK |
| MBP | Mouse IgG1 | 1 : 500 | MW | Boehringer-Mannheim, Mannheim, Germany |
| MAG | D7E10 | 1 : 4000 | MW | Dobersen et al., 1985 |
| MOG | 8–18C5 | 1 : 500 | MW | Linington et al., 1988 |
| CNPase | SMI91 | 1 : 2000 | MW | Sternberger Monoclonals, MD, USA |
| D-110 | D-110 | 1 : 5000 | MW | Lassmann et al., 2003 |
| HIF-1α | Clone 54 | 1 : 30 | MW | BD Transduction Laboratories, Lexington, KY, USA |
| hsp70 | C92F3A-5 | 1 : 200 | MW | StressGen, Victoria, Canada |
| CD3 | CD3-12 | 1 : 200 | MW | Serotec |
| CD20 | L 26 | 1 : 50 | MW | Dako, Glostrup, Denmark |
| CD68 | KP1 | 1 : 100 | MW | Dako, Glostrup, Denmark |
| iNOS | AB 5384 | 1 : 30.000 | MW | Chemicon, Temecula, USA |
| Caspase-3 | C92-603 | 1 : 5000 | MW | BD Biosciences, USA |
MW = microwave pretreatment.
Antibodies used for immunocytochemistry
| Antibody | Clone | Dilution | Pretreatment | Source |
|---|---|---|---|---|
| PLP | Rabbit pk | 1 : 1000 | MW | Serotec, Oxford, UK |
| MBP | Mouse IgG1 | 1 : 500 | MW | Boehringer-Mannheim, Mannheim, Germany |
| MAG | D7E10 | 1 : 4000 | MW | Dobersen et al., 1985 |
| MOG | 8–18C5 | 1 : 500 | MW | Linington et al., 1988 |
| CNPase | SMI91 | 1 : 2000 | MW | Sternberger Monoclonals, MD, USA |
| D-110 | D-110 | 1 : 5000 | MW | Lassmann et al., 2003 |
| HIF-1α | Clone 54 | 1 : 30 | MW | BD Transduction Laboratories, Lexington, KY, USA |
| hsp70 | C92F3A-5 | 1 : 200 | MW | StressGen, Victoria, Canada |
| CD3 | CD3-12 | 1 : 200 | MW | Serotec |
| CD20 | L 26 | 1 : 50 | MW | Dako, Glostrup, Denmark |
| CD68 | KP1 | 1 : 100 | MW | Dako, Glostrup, Denmark |
| iNOS | AB 5384 | 1 : 30.000 | MW | Chemicon, Temecula, USA |
| Caspase-3 | C92-603 | 1 : 5000 | MW | BD Biosciences, USA |
| Antibody | Clone | Dilution | Pretreatment | Source |
|---|---|---|---|---|
| PLP | Rabbit pk | 1 : 1000 | MW | Serotec, Oxford, UK |
| MBP | Mouse IgG1 | 1 : 500 | MW | Boehringer-Mannheim, Mannheim, Germany |
| MAG | D7E10 | 1 : 4000 | MW | Dobersen et al., 1985 |
| MOG | 8–18C5 | 1 : 500 | MW | Linington et al., 1988 |
| CNPase | SMI91 | 1 : 2000 | MW | Sternberger Monoclonals, MD, USA |
| D-110 | D-110 | 1 : 5000 | MW | Lassmann et al., 2003 |
| HIF-1α | Clone 54 | 1 : 30 | MW | BD Transduction Laboratories, Lexington, KY, USA |
| hsp70 | C92F3A-5 | 1 : 200 | MW | StressGen, Victoria, Canada |
| CD3 | CD3-12 | 1 : 200 | MW | Serotec |
| CD20 | L 26 | 1 : 50 | MW | Dako, Glostrup, Denmark |
| CD68 | KP1 | 1 : 100 | MW | Dako, Glostrup, Denmark |
| iNOS | AB 5384 | 1 : 30.000 | MW | Chemicon, Temecula, USA |
| Caspase-3 | C92-603 | 1 : 5000 | MW | BD Biosciences, USA |
MW = microwave pretreatment.
In situ hybridization
In situ hybridization for proteolipid protein (PLP) mRNA was performed on paraffin sections of the archival material as described in detail previously (Breitschopf et al., 1992; Lucchinetti et al., 1999). Hybridization was performed using digoxigenin-labelled riboprobes detected by alkaline phosphatase-conjugated anti-digoxigenin Fab fragments. For double staining after in situ hybridization, sections were incubated with anti-PLP antibody and processed as described above. Hybridization of the sections with sense probe was used as specificity control.
Detection of DNA fragmentation
For the detection of fragmented DNA, terminal deoxynucleotidyl transferase (TdT)-mediated incorporation of digoxigenin-labelled nucleotides was used as described previously (Stadelmann et al., 1998).
Results
General neuropathology of Baló-like lesions
Baló-like lesions were characterized either by a pattern of parallel rings with and without myelin or by a lamellar or mosaic pattern consisting of patches of demyelinated and myelinated areas surrounding a larger demyelinated area, the so-called storm centre (Fig. 1A). All these changes occurred on the background of an inflammatory reaction, mainly composed of T lymphocytes, macrophages and a variable number of CD20-positive B cells.
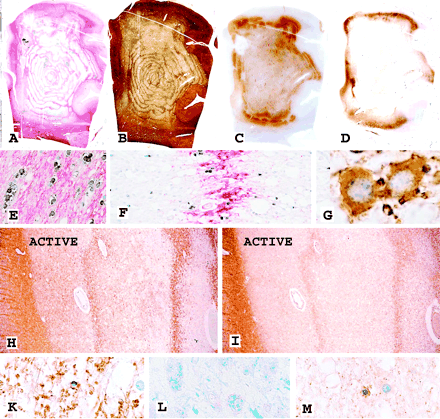
Large hemispheric brain lesion with concentric banding of myelinated and demyelinated tissue in a 38-year-old female patient diagnosed with multiple sclerosis 5 years ago with a clinical duration of the current illness of 2.5 weeks. (A) In situ hybridization for PLP mRNA (black) and immunocytochemistry for PLP protein (red) depicting the concentric nature of the lesion. Magnification ×1.3. (B) Bielschowsky silver impregnation for axons of the adjacent section shows massive loss of axons in the demyelinated, but preservation of axons in the myelinated areas resulting in an identical concentric layering of the lesion. Magnification ×1.3. (C) Immunocytochemistry for CD68 (macrophages) demonstrates macrophage accumulation at the outer edge of the lesion, representing the area of active demyelination. Magnification ×1.3. (D) Immunocytochemistry for the hypoxia-associated D-110 epitope reveals massive expression in a small rim just outside of the active lesion border. Magnification ×1.3. (E) In situ hybridization for PLP mRNA (black) and immunocytochemistry for PLP protein (red) shows normal density of oligodendrocytes in the periplaque white matter. Magnification ×150. (F) Same staining as in E demonstrates oligodendrocytes at high density in the areas of preserved myelin, but also some oligodendrocytes with PLP mRNA in the demyelinated layers. Magnification ×100. (G) Immunocytochemistry for myelin associated glycoprotein (MAG) shows intense intracytoplasmic reactivity in remyelinating oligodendrocytes in the center of the concentric lesion. Magnification ×1200. (H) The active edge of the concentric lesion reveals a zone of relative preservation of myelin oligodendrocyte glycoprotein (MOG) in the actively demyelinating area (ACTIVE), while MOG is lost in the completely demyelinated inner layers. Magnification ×20. (I) The adjacent section stained for MAG shows complete loss of this protein in the outer active areas (ACTIVE). The weak reactivity in the innermost demyelinated layer (right) is due to some remyelination. Magnification ×20. (K) Higher magnification of the active area, shown in H, stained for MOG. Note the partial preservation of myelin and an oligodendrocyte with nuclear condensation and margination, suggestive of apoptosis-like cell death. Magnification ×200. (L) Active area stained with Luxol fast blue. Nearly complete absence of myelin reactivity and presence of macrophages with granular myelin degradation products. Magnification ×400. (M) Same active area as shown in K, stained by immunocytochemistry for cyclic nucleotide phosphodiesterase (CNPase). CNPase reactivity is lost from myelin sheaths (seen in MOG staining in K). It is still preserved in an oligodendrocyte, which shows nuclear changes of apoptosis-like cell death. Magnification ×300.
The density of oligodendrocytes, identified by in situ hybridization for PLP mRNA and by immunocytochemistry for cyclic nucleotide phosphodiesterase (CNPase), was reduced in all areas of concentric lesions in comparison to the periplaque white matter (Fig. 1A, E, F), although the extent of oligodendrocyte loss varied between cases. The density of oligodendrocytes was invariably higher in the myelinated than in the demyelinated areas (Fig. 1F). In inactive lesions, both in the myelinated and demyelinated parts, oligodendrocytes revealed pronounced expression of PLP mRNA (Fig. 1F), and some of these cells also showed cytoplasmic reactivity for PLP and MAG protein (Fig. 1G). In particular, the demyelinated areas were associated with the appearance of thinly myelinated axons, suggesting remyelination. In comparison with their density in the periplaque white matter, axons were moderately reduced in the myelinated portions, but substantially lost in the demyelinated areas. Thus, even on gross examination a similar concentric lesion structure was evident in sections stained for axons and myelin (Fig. 1B).
Actively demyelinating lesions in Baló's concentric sclerosis show structural alterations suggestive of hypoxia-like tissue injury
In total, 21 concentric lesions were present in 14 patients, 19 of them in 12 patients revealed active demyelination at their edges (Table 2). Areas of lesional activity were defined by infiltration with macrophages (Fig. 1C) containing intracytoplasmic myelin degradation products reactive for all myelin proteins (Fig. 1L). Such active areas were restricted to the periphery of concentric lesions, suggesting radial plaque growth (Figs 1C and 3). The inner demyelinated rings either contained macrophages with empty vacuoles (neutral lipid stage of myelin degradation) or were devoid of macrophages. In these active areas, the myelin bands were sharply demarcated towards the inner layers of demyelinated tissue and rather blurred towards the outer layers of demyelination (Fig. 1H). The blurring corresponded to areas of ongoing myelin destruction.
Incidence of concentric demyelination in multiple sclerosis cases
| Concentric demyelination | Cases with active demyelination/total cases | Cases with active pattern III lesions | Cases with active lesions following other patterns of demyelination |
|---|---|---|---|
| Present | 12/14 | Acute MS: 8 | Acute MS: 0 |
| Chronic active MS: 4 | Chronic active MS: 0 | ||
| Absent | 18/28 | Acute MS: 1 | Acute MS: 5 |
| Chronic active MS: 0 | Chronic active MS: 12 |
| Concentric demyelination | Cases with active demyelination/total cases | Cases with active pattern III lesions | Cases with active lesions following other patterns of demyelination |
|---|---|---|---|
| Present | 12/14 | Acute MS: 8 | Acute MS: 0 |
| Chronic active MS: 4 | Chronic active MS: 0 | ||
| Absent | 18/28 | Acute MS: 1 | Acute MS: 5 |
| Chronic active MS: 0 | Chronic active MS: 12 |
MS = multiple sclerosis.
Incidence of concentric demyelination in multiple sclerosis cases
| Concentric demyelination | Cases with active demyelination/total cases | Cases with active pattern III lesions | Cases with active lesions following other patterns of demyelination |
|---|---|---|---|
| Present | 12/14 | Acute MS: 8 | Acute MS: 0 |
| Chronic active MS: 4 | Chronic active MS: 0 | ||
| Absent | 18/28 | Acute MS: 1 | Acute MS: 5 |
| Chronic active MS: 0 | Chronic active MS: 12 |
| Concentric demyelination | Cases with active demyelination/total cases | Cases with active pattern III lesions | Cases with active lesions following other patterns of demyelination |
|---|---|---|---|
| Present | 12/14 | Acute MS: 8 | Acute MS: 0 |
| Chronic active MS: 4 | Chronic active MS: 0 | ||
| Absent | 18/28 | Acute MS: 1 | Acute MS: 5 |
| Chronic active MS: 0 | Chronic active MS: 12 |
MS = multiple sclerosis.
Whenever active demyelination was present in patients with one or more concentric lesions, a uniform pattern of demyelination and tissue injury was observed (Table 2). This pattern consisted of complete loss of MAG, while other myelin proteins such as PLP and myelin oligodendrocyte glycoprotein (MOG) were initially well preserved (Fig. 1H, I, K). CNPase was also lost from myelin in actively demyelinating areas, but was still expressed in the oligodendrocyte cytoplasm (Fig. 1M). The differential loss of myelin proteins was associated with nuclear shrinkage, chromatin condensation and sometimes nuclear fragmentation in oligodendrocytes (Fig. 1K, M). Some of these cells were positive for DNA fragmentation, but did not react with antibodies to active caspase-3. Although some complement reactivity has been described in lesions with concentric demyelination before (Barnett and Prineas, 2004), with the technique applied in our study, none of the active concentric lesions showed reactivity for complement C9neo antigen on degenerating myelin or within myelin degradation products. Whether this discrepancy was due to methodological differences or to differences in the mechanisms of demyelination between cases is so far unresolved. The pattern of tissue damage described above resembles that found in early hypoxic/ischaemic white matter lesions (Aboul-Enein et al., 2003) and in a subset of active multiple sclerosis plaques, termed pattern III lesions (Lucchinetti et al., 2000; Aboul-Enein et al., 2003). Conversely, none of the active lesions in acute or chronic multiple sclerosis, which followed a different pattern of demyelination and tissue injury, showed concentric banding of myelin injury (Table 2).
All actively demyelinating areas as well as the inactive centre of the lesions showed profound perivascular cuffs, containing T-lymphocytes, some B-cells and macrophages. In addition, lymphocytes and macrophages diffusely infiltrated the lesions. While lymphocyte infiltration was similar in the active edge zones and the inactive centre of the lesions, macrophages were concentrated in the actively demyelinating areas (Fig. 1C). Macrophages in active lesional areas, but not in inactive zones, were highly reactive for inducible nitric oxide synthase (iNOS; Fig. 2A, B). iNOS reactivity was also present in activated microglia in a small zone of the periplaque white matter adjacent to the areas of active demyelination, although less pronounced than in macrophages in active areas (Figs 2A and 3).
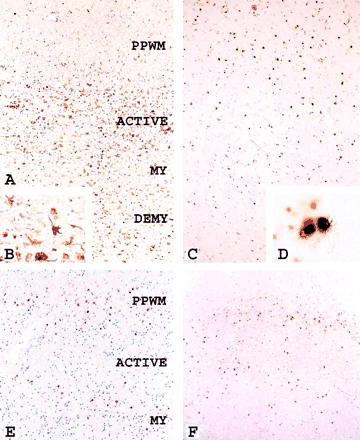
Actively demyelinating lesions in concentric sclerosis. (A) Expression of inducible nitric oxide synthase (iNOS) in the zone of active demyelination (ACTIVE) and to a much lower extent in the periplaque white matter (PPWM) or in the inner layers of myelinated or demyelinated tissue. Magnification ×50. (B) Higher magnification of A depicts iNOS expression in macrophages and microglia. Magnification ×500. (C) HIF-1α is mainly expressed in a small zone of the PPWM, just adjacent to the active edge of the lesion; the active area shows very little HIF-1α-expression, but the outermost layer of preserved myelin again shows a higher density of HIF-1α-positive cells. Magnification ×50. (D) Higher magnification of C demonstrating cytoplasmic expression and nuclear translocation of HIF-1α in small round cells resembling oligodendrocytes. Magnification ×800. (E) Expression of hsp70 in a concentric pattern comparable with that shown for HIF 1α in C. Magnification ×50. (F) A similar pattern of expression is also found for the hypoxia-associated epitope D-110. Magnification ×50.
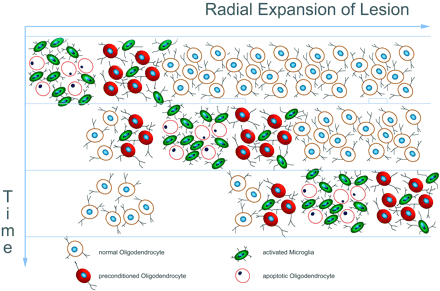
Schematic summary of neuropathological features of active Baló-like lesions. Active demyelination is present at the outer edge of the lesion and associated with profound iNOS expression in microglia (green cells). The expression of proteins involved in hypoxic preconditioning is located in a small zone outside the actively demyelinating area and in the outermost rim of preserved myelin and shows an inverse pattern compared to iNOS (red cells).
Molecules involved in tissue preconditioning are expressed at the outer lesion edge and in the outermost rims of preserved myelin in actively growing concentric lesions
Since the morphological pattern of demyelination in actively expanding concentric lesions closely resembled that found in hypoxic white matter lesions, we studied the expression of molecules involved in tissue preconditioning. In this study we concentrated on three molecules: the D-110 epitope, which has recently been defined as a very sensitive and specific marker for hypoxia-like tissue damage in ischaemic and inflammatory brain lesions (Lassmann et al., 2003), hypoxia-inducible factor 1α (HIF-1α), a key regulator in the induction of hypoxic preconditioning (Bergeron et al., 2000; Sharp et al., 2001), and heat shock protein 70 (hsp70), a stress protein involved in tissue protection against hypoxic injury (Nowak, 1985; Pelham, 1986; Christians et al., 2002). We found a profound expression of HIF-1α and D-110 in a rim of periplaque white matter adjacent to the edge of the expanding lesions (Fig. 1D). In addition, cells reactive for these molecules were also found occasionally in the outermost layers of preserved myelin, but were rare or absent in areas of active demyelination (Fig. 2C–F), in inactive lesions or in active plaques following other patterns of demyelination (Table 3). Equally, upregulation of hsp70 was found in a large number of cells at the edge of expanding concentric lesions and, less consistently, in the outermost rims of preserved myelin (Table 4). However, hsp70 could also be detected at the edge of active lesions following other patterns of demyelination in the absence of concentric banding (Table 3).
Density of cells with expression of HIF-1α, D-110 and hsp70 in active multiple sclerosis lesions
| HIF-1α | D-110 | Hsp70 | |
|---|---|---|---|
| Acute WM stroke | 225 ± 12 | 254 ± 47 | n.d. |
| MS pattern III | 252 ± 18 | 273 ± 58 | + to +++ |
| MS other patterns | 70 ± 15 | 48 ± 22 | + to +++ |
| HIF-1α | D-110 | Hsp70 | |
|---|---|---|---|
| Acute WM stroke | 225 ± 12 | 254 ± 47 | n.d. |
| MS pattern III | 252 ± 18 | 273 ± 58 | + to +++ |
| MS other patterns | 70 ± 15 | 48 ± 22 | + to +++ |
The expression of HIF-1α, D-110 and hsp70 was determined at the edge of actively expanding lesions; values for HIF-1a and D-110 are given as cells/mm2; for hsp70 a semiquantitative rating was performed: 0 = no cells stained; + = 1–10 cells/mm2; ++ = 11–100 cells/mm2; +++ = more than 100 cells/mm2. MS = multiple sclerosis; n.d. = not done.
Density of cells with expression of HIF-1α, D-110 and hsp70 in active multiple sclerosis lesions
| HIF-1α | D-110 | Hsp70 | |
|---|---|---|---|
| Acute WM stroke | 225 ± 12 | 254 ± 47 | n.d. |
| MS pattern III | 252 ± 18 | 273 ± 58 | + to +++ |
| MS other patterns | 70 ± 15 | 48 ± 22 | + to +++ |
| HIF-1α | D-110 | Hsp70 | |
|---|---|---|---|
| Acute WM stroke | 225 ± 12 | 254 ± 47 | n.d. |
| MS pattern III | 252 ± 18 | 273 ± 58 | + to +++ |
| MS other patterns | 70 ± 15 | 48 ± 22 | + to +++ |
The expression of HIF-1α, D-110 and hsp70 was determined at the edge of actively expanding lesions; values for HIF-1a and D-110 are given as cells/mm2; for hsp70 a semiquantitative rating was performed: 0 = no cells stained; + = 1–10 cells/mm2; ++ = 11–100 cells/mm2; +++ = more than 100 cells/mm2. MS = multiple sclerosis; n.d. = not done.
Expression of hsp70 in a concentric lesion of acute multiple sclerosis
| Area | Hsp 70 | CD68 |
|---|---|---|
| PPWM | 144 | 327 |
| DM1 (early active) | 36 | 726 |
| MY1 (inactive) | 131 | 379 |
| DM2 (early active) | 7 | 948 |
| MY2 (inactive) | 151 | 369 |
| DM3 (late active) | 26 | 798 |
| MY3 (inactive) | 62 | 387 |
| Centre (inactive) | 0 | 752 |
| Area | Hsp 70 | CD68 |
|---|---|---|
| PPWM | 144 | 327 |
| DM1 (early active) | 36 | 726 |
| MY1 (inactive) | 131 | 379 |
| DM2 (early active) | 7 | 948 |
| MY2 (inactive) | 151 | 369 |
| DM3 (late active) | 26 | 798 |
| MY3 (inactive) | 62 | 387 |
| Centre (inactive) | 0 | 752 |
The values for Hsp 70 and CD68 represent immunoreactive cells/mm2. The activity of the lesional areas was defined according to Brück et al. (1995). Early active = macrophages contain myelin degradation products reactive for all myelin proteins; late active = macrophages contain myelin degradation products, reactive only for MBP and PLP but not for MOG; inactive = macrophages contain neutral lipid degradation products but no reactivity for myelin proteins. Preferential expression of hsp70 is seen in the myelinated as opposed to the demyelinated layers of a concentric lesion.
Expression of hsp70 in a concentric lesion of acute multiple sclerosis
| Area | Hsp 70 | CD68 |
|---|---|---|
| PPWM | 144 | 327 |
| DM1 (early active) | 36 | 726 |
| MY1 (inactive) | 131 | 379 |
| DM2 (early active) | 7 | 948 |
| MY2 (inactive) | 151 | 369 |
| DM3 (late active) | 26 | 798 |
| MY3 (inactive) | 62 | 387 |
| Centre (inactive) | 0 | 752 |
| Area | Hsp 70 | CD68 |
|---|---|---|
| PPWM | 144 | 327 |
| DM1 (early active) | 36 | 726 |
| MY1 (inactive) | 131 | 379 |
| DM2 (early active) | 7 | 948 |
| MY2 (inactive) | 151 | 369 |
| DM3 (late active) | 26 | 798 |
| MY3 (inactive) | 62 | 387 |
| Centre (inactive) | 0 | 752 |
The values for Hsp 70 and CD68 represent immunoreactive cells/mm2. The activity of the lesional areas was defined according to Brück et al. (1995). Early active = macrophages contain myelin degradation products reactive for all myelin proteins; late active = macrophages contain myelin degradation products, reactive only for MBP and PLP but not for MOG; inactive = macrophages contain neutral lipid degradation products but no reactivity for myelin proteins. Preferential expression of hsp70 is seen in the myelinated as opposed to the demyelinated layers of a concentric lesion.
Expression of these molecules was not restricted to a single cell type, although it was most prominently found in oligodendrocytes (Fig. 2C–F) and to a lesser degree in astrocytes and microglia. In addition, neuronal expression of hypoxia-associated molecules was found in lesions involving the cerebral cortex. HIF-1α was not only present in the cytoplasm, but also translocated into the nucleus (Fig. 2D).
Discussion
Baló's concentric sclerosis is considered a variant of inflammatory demyelinating diseases clinically characterized by its acute onset, rapid progression and large lesions. With regard to pathology, the most conspicuous feature is the presence of large concentric lesions, consisting of alternating layers of preserved and destroyed myelin (Courville, 1970). Although large Baló-type lesions are very rare, small concentric bands of demyelinated and myelinated tissue are not infrequently found in a subset of fulminant active lesions, in particular in acute and less commonly in chronic multiple sclerosis (Barnett and Prineas, 2004). The pathophysiological mechanisms responsible for the formation of such lesions have up to now remained enigmatic. According to our current results the concentric layering of damaged and preserved tissue may be explained by tissue preconditioning in a steadily expanding lesion.
This interpretation is based on several key observations. First, whenever active demyelination and tissue destruction were present in concentric lesions, they followed a pattern of demyelination and tissue injury, which has recently been identified as a characteristic hallmark of hypoxia-like tissue damage (Aboul-Enein et al., 2003; Lassmann et al., 2003). The frequent association of concentric demyelination with this particular type of lesion has also recently been described in a study on early stages of multiple sclerosis lesions (Barnett and Prineas, 2004). The cardinal feature of this type of tissue injury is initial damage of the most distal (periaxonal) part of oligodendrocyte processes (Waxman et al., 1992), resulting in clumping and loss of MAG and CNPase within the myelin sheaths (Itoyama et al., 1980, 1982; Aboul-Enein et al., 2003). This is associated with caspase-independent apoptotic-like oligodendrocyte death (Aboul-Enein et al., 2003). Nuclear condensation and fragmentation in oligodendrocytes has previously been described in actively demyelinating Baló lesions (Yao et al., 1994). Furthermore, it has been suggested that hypoxia may contribute to the development of concentric lesions. This was based on the observation that concentric layering of intact and injured tissue is occasionally found in human white matter stroke lesions (Courville, 1970) and in experimental models of cyanide intoxication (Ferraro, 1933).
Second, we found a profound expression of molecules involved in tissue preconditioning in a small zone of periplaque white matter immediately adjacent to actively demyelinating concentric lesions. Expression of these molecules was also found in the outermost lamella of preserved myelin. Our results are supported by a recent study on gene expression in the normal-appearing white matter of multiple sclerosis patients, which, by using a microarray technology, showed that the mRNAs of multiple genes, clustered by their involvement in hypoxic preconditioning, are significantly upregulated in comparison with control white matter (Graumann et al., 2003).
HIF-1α is a central master switch in the induction of tissue preconditioning (Bergeron et al., 2000; Bernaudin et al., 2003). As a transcription factor, it regulates the expression of multiple downstream genes, which are involved in tissue protection following sublethal injury (Sharp et al., 2001; Sharp and Bernaudin, 2004). Hsp-70 acts as a chaperone involved in refolding of damaged intracellular proteins and exerts profound protective effects in hypoxic conditions (Christians et al., 2002). Being present at the edge of the lesions and in the layers of preserved myelin, which are adjacent to zones of active myelin destruction, these molecules may increase the resistance of the tissue against the noxious insult. Both HIF-1α (Sharp and Bernaudin, 2004) and hsp70 (D'Souza et al., 1994) are not only induced by hypoxia, but also by a variety of proinflammatory cytokines present in active multiple sclerosis lesions (Cannella and Raine, 1995; Mycko et al., 2003). Thus, not unexpectedly, hsp70 was expressed at the edge of all active multiple sclerosis lesions irrespective of the presence of concentric demyelination. However, in cases with concentric demyelination, hsp70 showed a concentric pattern of upregulation. In contrast, nuclear translocation of HIF-1α reactivity and D-110 expression were restricted to inflammatory demyelinating lesions following a pattern of tissue injury closely resembling that in acute white matter stroke lesions.
The question of what could cause hypoxia-like tissue injury in Baló's concentric sclerosis remains unanswered. Microvascular damage has been suggested in the original descriptions of Courville (Courville, 1970), and microvessel thrombosis can sometimes be observed in lesions of fulminant acute multiple sclerosis (Wakefield et al., 1994). Alternatively, a sort of histotoxic hypoxia may be induced by toxic inflammatory mediators through mitochondrial injury. Recent studies provide evidence for mitochondrial damage in multiple sclerosis lesions (Lu et al., 2000; Kalman and Leist, 2003; McDonough et al., 2003), which could be mediated by reactive oxygen (Lu et al., 2000) and nitric oxide species (Bolanos et al., 1997; Heales et al., 1999). The expression of iNOS within macrophages in actively demyelinating plaques and within microglia at the plaque edges in concentric lesions, as described here, supports the hypothesis that nitric oxide intermediates may be involved in the pathogenesis of these lesions. Although iNOS reactivity is found in macrophages also in non-concentric active as well as in more chronic multiple sclerosis lesions (Liu et al., 2001), the amount of oxygen and nitrogen intermediates produced in the latter may be lower compared to that in concentric plaques.
It has been suggested previously that concentric lesions may be formed by remyelination within the plaques (Moore et al., 1985). Indeed we found evidence for remyelinating oligodendrocytes within Baló-like lesions. These cells were, however, mainly present in inactive lesions and were not restricted to the zones of preserved myelin, but also seen in the demyelinated areas. As in other lesions of experimental inflammatory demyelination (Rodriguez et al., 1994) and of multiple sclerosis (Lucchinetti et al., 1999), the expression of mRNA for myelin proteins was nearly completely lost within the areas of active demyelination and even in the outer rims of preserved myelin located adjacent to the zones of active demyelination. This fact, together with the morphological evidence of thick myelin sheaths in the myelinated concentric tissue layers, argues against a role for remyelination in the formation of concentric lesions. However, since tissue preconditioning apparently does not specifically affect a certain cell type, oligodendrocyte progenitor cells may also be protected and produce new myelin in the preserved concentric rims when partial demyelination is present. We have previously shown that remyelination is sparse or absent in pattern III lesions of multiple sclerosis patients. Although some remyelination was present in most of the concentric lesions described here, it too was minor compared with that present in classical pattern II plaques, where extensive recruitment of oligodendrocytes and the formation of new myelin sheaths is a typical and prominent feature and occurs closely adjacent to the zone of active myelin destruction (Lucchinetti et al., 1999, 2000).
In conclusion, our studies offer a possible explanation for the formation of concentric lesions in Baló's concentric sclerosis (Fig. 3). The inflammatory process may induce an exaggerated activation of microglia and macrophages, resulting in extensive local production of nitric oxide intermediates and oxygen radicals. These molecules may impair mitochondrial function and provoke, through histotoxic hypoxia, the destruction of myelin sheaths and, less effectively, of axons. Like most multiple sclerosis lesions, concentric plaques grow by radial expansion around an inactive plaque core. At the edge of the expanding lesions sublethal tissue injury may induce the expression of molecules involved in tissue preconditioning. This appears to be a physiological response of the tissue to counteract radial plaque growth. However, in the case of aggressive lesion progression this zone of protected tissue may become run over, leading to new demyelination outside the protected zone. This may finally lead to the concentric patterning of demyelination and preserved myelin that is characteristic of Baló's disease.
We thank Marianne Leiszer and Helene Breitschopf for expert technical assistance. This study was funded by Fond zur Förderung der wissenschaftlichen Forschung, Austria (grant P 16063-B02), the US Multiple Sclerosis Society (grant RG 3051-A-1) and a prize from the Roman, Marga and Marielle Sobek Foundation. C.S. is supported by the Gemeinnützige Hertie-Stiftung and the medical faculty of the University of Göttingen (junior research group).
References
Aboul-Enein F, Rauschka H, Kornek B, et al. Preferential loss of myelin associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases.
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion.
Bergeron M, Gidday JM, Yu AY, et al. Role of hypoxia-inducible factor 1 in hypoxia-induced ischemic tolerance in neonatal rat brain.
Bernaudin M, Tang Y, Reilly M, et al. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance.
Bickler PE, Donohoe PH. Adaptive responses of vertebrate neurons to hypoxia.
Bolanos JP, Almeida A, Stewart V, et al. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases.
Breitschopf H, Suchanek G, Gould RM, et al. In situ hybridization with digoxigenin-labeled probes: sensitive and reliable detection method applied to myelinating rat brain.
Brück W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions.
Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions.
Chen CJ, Chu NS, Lu CS, Sung CY. Serial magnetic resonance imaging in patients with Baló's concentric sclerosis: natural history of lesion development.
Christians ES, Yan LJ, Benjamin IJ. Heat shock factor-1 and heat shock proteins: critical partners in protection against acute cell injury.
Courville CB. Concentric sclerosis. In: Vinken PJ, Bruyn GW, editors. Multiple sclerosis and other demyelinating diseases. Amsterdam: North-Holland;
D'Souza SD, Antel JP, Freedman MS. Cytokine induction of heat shock protein expression in human oligodendrocytes: an interleukin-1 mediated mechanism.
Dobersen MJ, Hammer JA, Noronha AB, et al. Generation and characterization of mouse monoclonal antibodies to myelin associated glycoprotein (MAG).
Ferraro A. Experimental toxic encephalopathy. Diffuse sclerosis following subcutaneous injection of potassium-cyanide.
Garbern J, Spence AM, Alvord EC Jr. Baló's concentric demyelination diagnosed premortem.
Gharagozloo AM, Poe LB, Collins GH. Antemortem diagnosis of Baló concentric sclerosis: correlative MR imaging and pathologic features.
Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult.
Hallervorden J, Spatz H. Über die konzentrische Sklerose und die physikalisch-chemischen Faktoren bei der Ausbreitung von Entmarkungsprozessen.
Hanemann CO, Kleinschmidt A, Reifenberger G, Freund H-J, Seitz RJ. Baló's concentric sclerosis followed by MRI and positron emission tomography.
Heales SJ, Bolanos JP, Stewart VC, et al. Nitric oxide, mitochondria and neurological disease.
Itoyama Y, Sternberger NH, Webster HD, et al. Immunocytochemical observation on the distribution of myelin-associated glycoprotein and myelin basic protein in multiple sclerosis lesions.
Itoyama Y, Webster HDF, Sternberger NH, et al. Distribution of papovavirus, myelin-associated glycoprotein and myelin basic protein in progressive multifocal leukoencephalopathy lesions.
Kalman B, Leist TP. A mitochondrial component of neurodegeneration in multiple sclerosis.
Karaarslan E, Altintas A, Senol U, et al. Baló's concentric sclerosis: clinical and radiological features of five cases.
Korte JH, Bom EP, Vos LD, Breuer TJM, Wondergem JHM. Baló concentric sclerosis: MR diagnosis.
Kuroiwa Y. Concentric sclerosis. In: Koetsier JC, editor. Demyelinating diseases. Amsterdam: Elsevier Science Publishers;
Lassmann H, Reindl M, Rauschka H, et al. A new paraclinical CSF marker for hypoxia-like tissue damage in multiple sclerosis lesions.
Linington C, Bradl M, Lassmann H, et al. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating antibodies against a myelin oligodendrocyte glycoprotein.
Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrothyrosine in multiple sclerosis lesions.
Lu F, Selak M, O'Connor J, Croul S, Lorenzana C, Butuoni C, Kalman B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis.
Lucchinetti C, Brück W, Parisi J, et al. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 117 cases.
Lucchinetti CF, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination.
Marburg O. Die sogenannte ‘akute multiple Sklerose’ (Encephalomyelitis periaxialis scleroticans).
McDonough J, Dutta R, Gudz T, et al. Decreases in GABA and mitochondrial genes are implicated in MS cortical pathology through microarray analysis of postmortem MS cortex.
Miyata K, Itoyama Y, Kobayashi T, Yasumori K, Goto I. A case of demyelinating disease showing a peculiar honeycomb-like and lamellar structure on magnetic resonance imaging.
Moore GRW, Neumann PE, Suzuki K, Lijtmaer HN, Traugott U, Raine CS. Baló's concentric sclerosis: new observations on lesion development.
Mycko MP, Papoian R, Boschert U, Raine CS, Selmaj KW. cDNA microarray analysis in multiple sclerosis lesions: detection of genes associated with disease activity.
Ng SH, Ko SF, Cheung YC, et al. MRI features of Baló's concentric sclerosis.
Nowak TS. Synthesis of a stress protein following transient ischemia in the gerbil.
Pelham HR. Speculations on the functions of the major heat shock and glucose regulated proteins.
Revel MP, Valiente E, Gray F, et al. Concentric MR patterns in multiple sclerosis. Report of two cases.
Rodriguez M, Prayoonwiwat N, Howe C, Sanborn K. Proteolipid protein gene expression in demyelination and remyelination of the central nervous system: a model for multiple sclerosis.
Sharp FR, Bergeron M, Bernaudin M. Hypoxia-inducible factor in brain.
Spiegel M, Krüger H, Hofmann E, Kappos L. MRI study of Baló's concentric sclerosis before and after immunosuppressive therapy.
Stadelmann C, Bruck W, Bancher C, et al. Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis.
Vass K, Lassmann H, Wekerle H, Wisniewski HM. The distribution of Ia-antigen in the lesion of rat acute experimental allergic encephalomyelitis.
Vass K, Welch JW, Nowak ST. Localization of 70kD stress protein induction in gerbil brain after ischemia.
Wakefield AJ, More LJ, Difford J, McLaughlin JE. Immunohistochemical study of vascular injury in acute multiple sclerosis.
Waxman SG, Black JA, Stys PK, Ransom BR. Ultrastructural concomitants of anoxic injury and early post-anoxic recovery in rat optic nerve.
Author notes
1Brain Research Center, Medical University of Vienna, Vienna, Austria, 2Department of Neuropathology, University of Goettingen, Goettingen, Germany, 3Department of Neuropathology, Queen's University, Kingston, Ontario, Canada, 4National Institute for Longevity Sciences, NCGG, Aichi, Japan, 5Department of Neurology, Municipal Hospital, Szekesfehervar 6Department of Neuropathology, Municipal Hospital, Esztergom, Hungary, 7Department of Neurology, Mayo Clinic, Rochester, MN, USA and 8Department of Neuropsychiatry, Santo Tomas University, Manila, Philippines


{kind=link}
{kind=link}
{kind=link}