




Abstract
The rate of brain atrophy and its relationship to clinical disease progression in progressive supranuclear palsy (PSP) and multiple system atrophy (MSA) is not clear. Twenty-four patients with PSP, 11 with MSA-P (Parkinsonian variant), 12 with Parkinson's disease, and 18 healthy control subjects were recruited for serial MRI scans, clinical assessments and formal neuropsychological evaluations in order to measure brain atrophy during life and its association with disease progression in PSP and MSA-P. Serial scans were registered and rates of whole brain atrophy calculated from the brain-boundary shift integral. Regional rates of atrophy were calculated in the brainstem (midbrain and pons), the cerebellum, the lateral and third ventricles as well as frontal and posterior inferior brain regions, by locally registering to a region of interest in order to derive a local boundary shift integral (BSI). 82% of recruited subjects completed serial MRI scans (17 PSP, 9 MSA-P, 9 Parkinson's disease patients and 18 healthy controls). Mean (SD) annualized rates of whole-brain atrophy were greatest in PSP: 1.2% (1.0%), three times that in controls. Mean (SD) midbrain atrophy rates in PSP, 2.2% (1.5%), were seven times greater than in healthy controls. In MSA-P, atrophy rates were greatest in the pons: 4.5% (3.2%), over 20 times that in controls and three times the rate of pontine atrophy in PSP. Atrophy rates in Parkinson's disease were not significantly different from control rates of atrophy. Variability in the atrophy rates was lower when calculated using the BSI rather than manual measurements. Worsening motor deficit was associated with midbrain atrophy in PSP, and ponto-cerebellar atrophy in MSA-P. Worsening executive dysfunction was associated with increased rates of frontal atrophy in PSP. Cerebellar atrophy rates were better discriminators of MSA-P than cross-sectional volumes. We confirm that serial MRI can be applied to measure whole brain and regional atrophy rates in PSP and MSA-P. Regional rather than whole-brain atrophy rates better discriminate PSP and MSA-P from healthy controls. Clinico-radiological associations suggest these regional atrophy rates have potential as markers of disease progression in trials of novel therapies.
Introduction
Progressive supranuclear palsy (PSP) (Steele et al., 1964) and multiple system atrophy (MSA) (Graham and Oppenheimer, 1969) are both neurodegenerative disorders. Recent studies have suggested that PSP is more common than often considered with a prevalence of around 5 per 100000 (Nath et al., 2001), with a more parkinsonian PSP phenotype contributing to the difficulty of early clinical diagnosis (Williams et al., 2005).
There has been interest in the clinico-radiological correlates of PSP and MSA for many years. However, so far, imaging studies have been largely cross sectional. Some have been observational and subjective (Stern et al., 1989), others have employed quantitative linear measurements (Schrag et al., 2000; Asato et al., 2000). Most recently, volumetric region of interest (ROI) studies, voxel-based morphometry and ratios of regional area measurements, have been used to demonstrate structural differences between these diseases (Cordato et al., 2002; Brenneis et al., 2003; Groschel et al., 2004; Cordato et al., 2005; Oba et al., 2005; Paviour et al., 2005b). All of these cross-sectional studies are limited by between-subject differences and by definition can only imply that group differences reflect that atrophy has occurred. To calculate rate of brain atrophy using MRI, serial imaging is necessary. Serial imaging studies in MSA using qualitative and quantitative area measurements have been undertaken (Horimoto et al., 2000; Horimoto et al., 2002; Konagaya et al., 2002; Watanabe et al., 2002) and registration of longitudinal volumetric MRI brain scans has been extensively evaluated in Alzheimer's disease (Fox et al., 1996; Fox and Freeborough, 1997; Fox et al., 1999; Jack et al., 2000) and fronto-temporal dementia (Chan et al., 2001a; Chan et al., 2001b; Schott et al., 2003). In PSP, rates of whole brain and ventricular atrophy have recently been measured using serial MRI in a small group of patients (Josephs et al., 2006). However measuring regional atrophy rates using this technique has not been applied to PSP or MSA-P until now, and the use of MRI derived atrophy rates as markers of disease progression can only be confirmed if a meaningful clinical association is established.
The aims of this study were: (i) to determine rates of whole brain and regional brain atrophy in PSP, MSA-P, Parkinson's disease patients and healthy controls using registration of serial MRI brain scans; (ii) to determine whether regional brain atrophy rates differentiate PSP and MSA-P from each other and from Parkinson's disease patients and healthy controls; and (iii) to study the relationship between changes in disease severity and atrophy rates in PSP and MSA-P in order to help evaluate the potential use of atrophy rates as outcome measures in clinical trials.
Methods
Patients
We recruited patients with a clinical diagnosis of PSP, MSA-P (parkinsonian phenotype) or Parkinson's disease from a specialist movement disorders clinic. Patients were included if they fulfilled the NINDS-SPSP diagnostic criteria for clinically definite or clinically probable PSP (Litvan et al., 2003), the consensus criteria for clinical diagnosis of probable MSA (Gilman et al., 1999) or the Queen Square Brain Bank clinical diagnostic criteria for Parkinson's disease (Gibb and Lees, 1988). Whenever possible, spouses or partners of patients were recruited as healthy controls. Informed consent for inclusion in the study was obtained from all subjects and the study approved by the regional ethics committee at the National Hospital for Neurology and Neurosurgery. All patients included had a full clinical work up to exclude alternative diagnoses where appropriate. Clinical details and cross-sectional data from some of these subjects have been published previously (Price et al., 2004; Paviour et al., 2005b).
Clinical assessments
Scores on the Unified Parkinson's Disease Rating Scale (UPDRS) (Fahn et al., 1987) and the Hoehn and Yahr scale (Hoehn and Yahr, 1967) were recorded for each subject. Falls and postural instability as recorded in the UPDRS were noted, as was speech and swallowing difficulty. The Folstein Mini-Mental State Examination (MMSE) (Folstein et al., 1975) and the frontal assessment battery (FAB) (Dubois et al., 2000) were recorded at the bedside in PSP, MSA-P and Parkinson's disease patients.
Neuropsychological assessment
The neuropsychological assessment consisted of the following tests: the Mattis Dementia Rating Scale-2 (DRS-2) (which consists of the following subtests: attention, verbal and non-verbal memory, motor initiation, construction and conceptual ability) (Mattis, 1973); current verbal IQ based on completion of the Vocabulary, Similarities and Digit Span subtests of the revised Wechsler Adult Intelligence Scale (Wechsler, 1981); the Rey Auditory Verbal Learning Test (Rey, 1958); the Short Recognition Memory for Faces (Warrington, 1984); the Reitan Trail Making Test including Trail A, and Trail B (Reitan, 1958). In addition, patients were administered the Wisconsin Card Sorting Test (WCST) (Nelson, 1976); the semantic, phonemic and alternating semantic versions of the verbal fluency test (Benton, 1968) and the Paced Auditory Serial Addition Test (PASAT) (Gronwall and Wrightson, 1981). An apathy scale (Marin, 1990), and the Beck Anxiety and Depression Inventories (Beck et al., 1961; Beck and Steer, 1990), were completed by all patients. The pre-morbid level of intellectual functioning was estimated using the National Adult Reading Test (Nelson, 1982).
All patients prescribed levodopa, underwent clinical and neuropsychological assessments in the ‘on’ state with regard to their levodopa response. Neuropsychology assessments were undertaken on the same day that MRI scans were acquired in all except four patients in whom assessments were undertaken within 4 weeks of the scan date.
MRI
Patients and healthy controls underwent serial MRI. All T1-weighted volumetric magnetic resonance (MR) scans were acquired on the same 1.5 GE Signa Unit (General Electric, Milwaukee, WI, USA) using a spoiled gradient-echo technique (256 × 256 matrix, FOV 24 × 18 cm, TI/TR/TE/NEX/FLIP = 650 ms/13 ms/5.4 ms/1 ms/35°). This yielded 124 contiguous 1.5 mm thick slices. T2- and proton density-weighted scans were obtained in order to help exclude alternate pathologies that might have contributed to the clinical picture.
The digitized structural MR images were transferred to a SUN workstation (Sun Microsystems Inc, Mountain View, CA) and image analysis undertaken using the MIDAS (Medical Image Display and Analysis Software) package (Freeborough et al., 1997). Image analysis requires consistency in image acquisition and is affected by problems such as movement artefact or intensity inhomogeneity (‘shading’) artefact. For this study, post-acquisition correction of heterogeneity artefact was applied (Lewis and Fox, 2004). Images were registered to standard space, to ensure they were oriented in the same plane. Total intracranial volumes were measured for each scan (Whitwell et al., 2001). Whole brain volumes were calculated using the MIDAS software tool. All segmentations used intensity thresholding with thresholds set empirically as fixed fractions of mean brain intensity.
Region of interest segmentation protocols
A semi-automated seed intensity-threshold based processing routine was used to delineate regions that were ‘brain’ and ‘non-brain’ in each image using the MIDAS software tool to ‘plant’ a seed within the ROI on each scan slice. With the intensity threshold set at a predetermined limit, the brain/CSF boundary can be delineated without manual outlining. Each scan slice was reviewed and adjustments made to ensure accuracy of the measurements.
MR images were segmented in this way as scan pairs (baseline and follow-up) with the operator blinded to the identity, diagnosis and time sequence of the scans. Whole brain volumes were measured in order to calculate global cerebral atrophy. Lateral ventricle, brainstem (midbrain and pons), cerebellar, third ventricle, posterior inferior and frontal brain volumes were measured, as pathological and cross sectional imaging studies have demonstrated involvement of these regions as well as clinical associations in PSP and MSA-P. Protocols for segmentation of the whole brain and lateral ventricles have been described in detail elsewhere (Freeborough et al., 1997; Dalton et al., 2002). The superior cerebellar peduncle (SCP) was segmented using a protocol modified from a previous study (Paviour et al., 2005b). For all other ROI analysis, the operator measured the volume of the structure with two orthogonal views available. Methods and examples of midbrain, pontine and cerebellar volumes are shown in Fig. 1. For segmentation of the third ventricle, care was taken to exclude voxels within the aqueduct of Sylvius and within the lateral ventricles. To improve repeatability all voxels above the inferior border of the corpus callosum were excluded. An example of segmented third ventricle and lateral ventricle volume is shown in Fig. 2.
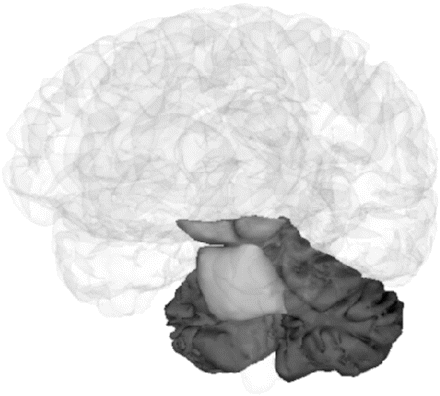
Regional segmentation demonstrating midbrain, pons and cerebellar volumes. For all ROI analysis, tissue boundaries were defined using the MIDAS software tool (Freeborough et al., 1997) with two orthogonal views available. Midbrain: The superior cut-off was taken as the upper border of the midbrain tegmentum in the mid-sagittal slice and the inferior border at the superior border of the pons in the mid sagittal slice. The posterior and anterior borders were defined by the brain tissue/CSF boundary (interpeduncular cistern anteriorly). Care was taken to include the quadrigeminal plate. Pons: The superior border was taken as a horizontal line extending posteriorly from the superior pontine notch. The inferior border was taken at the level of the inferior pontine notch. The anterior and lateral borders are clearly defined by CSF and the posterior border by the fourth ventricle. Cerebellum: segmentation included tissue in the cerebellar peduncles where it clearly connected with the cerebellar hemispheres. The anterior cut-off is taken as the most anterior slice to include cerebellar tissue. In all cases, care was taken to exclude all non-brain tissue.
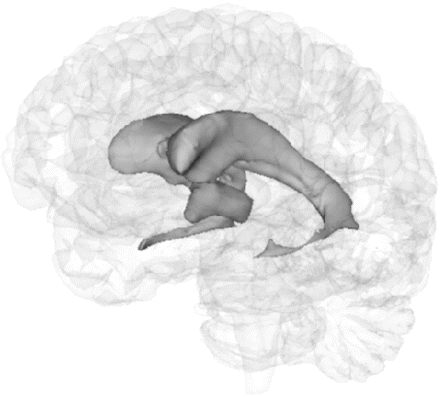
Regional segmentation demonstrating lateral and third ventricle volumes. Third ventricle: the anterior border of the ventricle was placed at the anterior commissure in the mid-sagittal section and the posterior border as the splenium of the corpus callosum. Care was taken to exclude voxels within the aqueduct of Sylvius. Lateral ventricle: a protocol for segmentation of the lateral ventricle has been described in detail previously (Dalton et al., 2002).
To evaluate the intrarater reproducibility, the same rater blindly repeated 10 randomly selected midbrain, pons, cerebellar and third ventricle measurements.
A template based, segmentation protocol was utilized to delineate frontal quadrants and posterior-inferior regions. The methods for frontal quadrant segmentation have been described previously (Chan et al., 2001a).
Atrophy rate calculation
For each scan pair, approximate change in brain and regional volume was calculated by subtracting the volume of the second segmented scan from the volume of the first segmented scan. The change in volume was also calculated directly and more precisely with the brain-boundary shift integral derived from positionally matched (registered) scans (Freeborough and Fox, 1997). Serial brain scans were first registered (spatially matched using 9 degrees of freedom) applying whole brain to whole brain matching (Woods et al., 1998).
Local boundary shift integrals (BSIs) were then measured by restricting the BSI calculation to the defined ROI. Briefly, serial brain images were registered applying whole brain to whole brain matching using a rigid-body registration as above. ROI were then further spatially matched by registering the baseline ROI to the follow up ROI using a six degrees of freedom registration. The region created by subtracting an eroded (by one voxel) intersection region from a dilated (by one voxel) union region of the resulting two brain regions was generated and a dilated (by one voxel) baseline and follow up ROI used to mask this in order to allow calculation of the BSI in this anatomical area of interest. This is illustrated schematically in Fig. 3. For the cerebellum, only the baseline cerebellar volume was manually segmented and the regional BSI calculated using only this baseline (dilated by one voxel) region. This technique has been applied to hippocampal ROI measurements in Alzheimer's disease (Barnes et al., 2004).
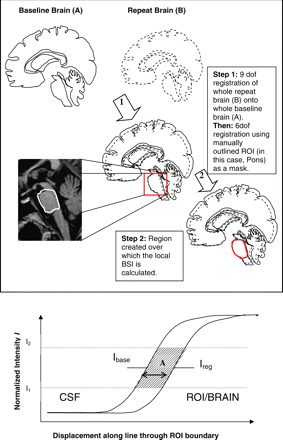
Top: summary of steps necessary to generate regional BSI (for the pons in this case). The steps leading to region of interest (ROI) registration are described. Firstly, a 9 degrees of freedom (dof) registration was used to match whole brains, then a 6 dof registration was applied to match ROI. Following this, the region that contained the ROI boundary shift was generated and the ROI BSI calculated in this region only. Bottom: Example of an idealised one-dimensional boundary shift. Area A is divided by the span of the intensity window I1-I2 to give the approximate distance over which the boundary has shifted, between Ibase and Ireg. This can be extended to three dimensions to give the volume loss over time.
Annualized changes were calculated by taking the difference between baseline and follow up measurements, and the BSIs in each group and adjusting for scan interval. Atrophy rates were calculated as a percentage change from the baseline volume.
Data were analysed using STATA version 8 (Stata Corporation, College station, TX, USA).
Clinical information (age, disease duration, UPDRS scores, Hoehn and Yahr score, MMSE and FAB scores) was compared using a one-way analysis of variance. Where statistically significant differences were noted, post hoc Bonferroni analysis was performed.
A one-way analysis of variance (ANOVA) test was used to compare whole brain and regional atrophy rates for normally distributed data, with post hoc Bonferroni tests where significant group differences were observed. A Mann–Whitney U-test was used to compare atrophy rates for non-parametric data.
Pitman's test was used to assess whether atrophy rates calculated using the brain-BSI and regional BSIs had lower variance than atrophy rates calculated from manually subtracted baseline and follow up measurements.
Logistic regression models were applied to assess the ability of BSI derived atrophy rates to discriminate between PSP and MSA-P.
For normally distributed data, linear regression was used to investigate the relationship between BSI derived whole brain and regional atrophy rates and change in clinical and neuropsychological test scores over time in the PSP and MSA-P groups separately. For this analysis the atrophy rate was used as the dependent variable with change in the clinical or neuropsychological test score as the independent variable. Age, disease duration and the baseline clinical test score were used as covariates in the analysis. Partial correlation coefficients (Corr.) were calculated to estimate the strength of the relationship if age and disease duration were constant. For non-normally distributed data, linear regression models with 95% bootstrapped confidence intervals (and implied P-values) were used to investigate the relationship between atrophy rates and change in test scores, with age, disease duration and the baseline clinical test as covariates. All 12 neuropsychological tests and the UPDRS II, III, Hoehn and Yahr and the FAB were assessed.
In order to investigate whether regions were independently associated with change in the clinical scores of motor deficit (UPDRS and Hoehn and Yahr) and bedside tests of cognition (FAB and MMSE), multiple regression models were performed with regional atrophy rate as the dependent variable and change in clinical score as the independent variable. Age, disease duration, baseline clinical rating scale score and the next most significant regional atrophy rate were included as covariates.
Results
Twenty-four patients with PSP, eleven with MSA-P, twelve with Parkinson's disease and eighteen healthy controls were recruited and had an initial scan. Three of the PSP patients were excluded from further follow up as the first scan was of poor quality due to excessive movement artefact. Two patients (one with PSP and one with MSA-P) were too unwell to attend for a second scan, two Parkinson's disease patients withdrew from the study and one with MSA-P and one with PSP died. One individual classified as PSP at the initial visit developed visual hallucinations and a clinical syndrome more in keeping with a clinical diagnosis of dementia with Lewy bodies and was excluded from data analysis. Follow up scans in one patient with PSP and one with Parkinson's disease were of poor quality and were not included in data analysis. Hence of 65 individuals recruited, 53 (17 PSP, 9 MSA-P, 9 Parkinson's disease patients and 18 healthy controls), or 82% had serial MRI scans analysed. In total, 33 of 35 patients completed baseline and follow up neuropsychological assessments. The results of neuropsychological assessments in these patients have been published previously (Paviour et al., 2005c). Four subjects with serial MRI scans have subsequently died and three who had consented to brain donation have had the clinical diagnosis confirmed at post-mortem (two with PSP, one with MSA-P).
Clinical data reported in Table 1.
Clinical features: mean (SD) range
| PSP | MSA-P | PD | Control | P-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ||||||
| Age (years) | 65.5 (5.6) 55–76 | 62.4 (8.1) 51–80 | 64.4 (8.3) 53–74 | 66.8 (5.4) 56–74 | 0.4 | |||||
| Disease duration (years) | 4.6 (1.6) 2–8 | 5.4 (1.7) 3–8 | 12.9 (4.3)a 7–20 | – | <0.001 | |||||
| Scan interval (days) | 292 (59)182–362 | 262 (66) 203–364 | 268 (52) 210–359 | 252 (40) 182–328 | 0.2 | |||||
| UPDRSII | ||||||||||
| Time1 | 18.4 (5.9) | 24.1 (7.6)b | 13.4 (4.6) | – | 0.003 | |||||
| Time2 | 27.8 (7.9) | 30.4 (7.6) | 18.4 (5.8)c | – | 0.003 | |||||
| UPDRSIII | ||||||||||
| Time1 | 18.5 (6.3) | 24.7 (9.5) | 16.4 (3.1)d | – | 0.03 | |||||
| Time2 | 28.5 (7.8) | 32.0 (10.3) | 21.3 (7.2)e | – | 0.03 | |||||
| HY | ||||||||||
| Time1 | 3.4 (0.6) | 3.8 (0.8) | 2.7 (0.5)f | – | 0.003 | |||||
| Time2 | 4.1 (0.7) | 4.2 (0.8) | 3.1 (0.6)g | – | 0.003 | |||||
| MMSE | ||||||||||
| Time1 | 26.6 (2.6) | 25.9 (3.4) | 28.2 (2.5) | 29.5 (0.6)h | <0.001 | |||||
| Time2 | 26.2 (3.1) | 26.4 (3.4) | 27.8 (2.3) | |||||||
| FAB | ||||||||||
| Time1 | 12.6 (2.8) | 14.6 (2.8) | 17.0 (1.1)i | – | <0.001 | |||||
| Time2 | 10.5 (4.4) | 13.2 (3.3) | 16.1 (2.4)j | – | 0.003 | |||||
| PSP | MSA-P | PD | Control | P-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ||||||
| Age (years) | 65.5 (5.6) 55–76 | 62.4 (8.1) 51–80 | 64.4 (8.3) 53–74 | 66.8 (5.4) 56–74 | 0.4 | |||||
| Disease duration (years) | 4.6 (1.6) 2–8 | 5.4 (1.7) 3–8 | 12.9 (4.3)a 7–20 | – | <0.001 | |||||
| Scan interval (days) | 292 (59)182–362 | 262 (66) 203–364 | 268 (52) 210–359 | 252 (40) 182–328 | 0.2 | |||||
| UPDRSII | ||||||||||
| Time1 | 18.4 (5.9) | 24.1 (7.6)b | 13.4 (4.6) | – | 0.003 | |||||
| Time2 | 27.8 (7.9) | 30.4 (7.6) | 18.4 (5.8)c | – | 0.003 | |||||
| UPDRSIII | ||||||||||
| Time1 | 18.5 (6.3) | 24.7 (9.5) | 16.4 (3.1)d | – | 0.03 | |||||
| Time2 | 28.5 (7.8) | 32.0 (10.3) | 21.3 (7.2)e | – | 0.03 | |||||
| HY | ||||||||||
| Time1 | 3.4 (0.6) | 3.8 (0.8) | 2.7 (0.5)f | – | 0.003 | |||||
| Time2 | 4.1 (0.7) | 4.2 (0.8) | 3.1 (0.6)g | – | 0.003 | |||||
| MMSE | ||||||||||
| Time1 | 26.6 (2.6) | 25.9 (3.4) | 28.2 (2.5) | 29.5 (0.6)h | <0.001 | |||||
| Time2 | 26.2 (3.1) | 26.4 (3.4) | 27.8 (2.3) | |||||||
| FAB | ||||||||||
| Time1 | 12.6 (2.8) | 14.6 (2.8) | 17.0 (1.1)i | – | <0.001 | |||||
| Time2 | 10.5 (4.4) | 13.2 (3.3) | 16.1 (2.4)j | – | 0.003 | |||||
Post hoc analyses: aPD versus PSP/MSA-P, P < 0.001; bPD versus MSA-P, P = 0.002; cPD versus MSA-P, P = 0.004, PD versus PSP, P = 0.01; dPD versus MSA-P, P = 0.04; ePD versus MSA-P, P = 0.03; fPD versus PSP, P = 0.05, PD versus MSA-P, P = 0.003; gPD versus PSP, P = 0.007, PD versus MSA-P, P = 0.005; hcontrol versus PSP, P = 0.004, control versus MSA-P, P = 0.003; iPD versus PSP, P < 0.001; jPD versus PSP, P = 0.003; PD = Parkinson's disease.
Clinical features: mean (SD) range
| PSP | MSA-P | PD | Control | P-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ||||||
| Age (years) | 65.5 (5.6) 55–76 | 62.4 (8.1) 51–80 | 64.4 (8.3) 53–74 | 66.8 (5.4) 56–74 | 0.4 | |||||
| Disease duration (years) | 4.6 (1.6) 2–8 | 5.4 (1.7) 3–8 | 12.9 (4.3)a 7–20 | – | <0.001 | |||||
| Scan interval (days) | 292 (59)182–362 | 262 (66) 203–364 | 268 (52) 210–359 | 252 (40) 182–328 | 0.2 | |||||
| UPDRSII | ||||||||||
| Time1 | 18.4 (5.9) | 24.1 (7.6)b | 13.4 (4.6) | – | 0.003 | |||||
| Time2 | 27.8 (7.9) | 30.4 (7.6) | 18.4 (5.8)c | – | 0.003 | |||||
| UPDRSIII | ||||||||||
| Time1 | 18.5 (6.3) | 24.7 (9.5) | 16.4 (3.1)d | – | 0.03 | |||||
| Time2 | 28.5 (7.8) | 32.0 (10.3) | 21.3 (7.2)e | – | 0.03 | |||||
| HY | ||||||||||
| Time1 | 3.4 (0.6) | 3.8 (0.8) | 2.7 (0.5)f | – | 0.003 | |||||
| Time2 | 4.1 (0.7) | 4.2 (0.8) | 3.1 (0.6)g | – | 0.003 | |||||
| MMSE | ||||||||||
| Time1 | 26.6 (2.6) | 25.9 (3.4) | 28.2 (2.5) | 29.5 (0.6)h | <0.001 | |||||
| Time2 | 26.2 (3.1) | 26.4 (3.4) | 27.8 (2.3) | |||||||
| FAB | ||||||||||
| Time1 | 12.6 (2.8) | 14.6 (2.8) | 17.0 (1.1)i | – | <0.001 | |||||
| Time2 | 10.5 (4.4) | 13.2 (3.3) | 16.1 (2.4)j | – | 0.003 | |||||
| PSP | MSA-P | PD | Control | P-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ||||||
| Age (years) | 65.5 (5.6) 55–76 | 62.4 (8.1) 51–80 | 64.4 (8.3) 53–74 | 66.8 (5.4) 56–74 | 0.4 | |||||
| Disease duration (years) | 4.6 (1.6) 2–8 | 5.4 (1.7) 3–8 | 12.9 (4.3)a 7–20 | – | <0.001 | |||||
| Scan interval (days) | 292 (59)182–362 | 262 (66) 203–364 | 268 (52) 210–359 | 252 (40) 182–328 | 0.2 | |||||
| UPDRSII | ||||||||||
| Time1 | 18.4 (5.9) | 24.1 (7.6)b | 13.4 (4.6) | – | 0.003 | |||||
| Time2 | 27.8 (7.9) | 30.4 (7.6) | 18.4 (5.8)c | – | 0.003 | |||||
| UPDRSIII | ||||||||||
| Time1 | 18.5 (6.3) | 24.7 (9.5) | 16.4 (3.1)d | – | 0.03 | |||||
| Time2 | 28.5 (7.8) | 32.0 (10.3) | 21.3 (7.2)e | – | 0.03 | |||||
| HY | ||||||||||
| Time1 | 3.4 (0.6) | 3.8 (0.8) | 2.7 (0.5)f | – | 0.003 | |||||
| Time2 | 4.1 (0.7) | 4.2 (0.8) | 3.1 (0.6)g | – | 0.003 | |||||
| MMSE | ||||||||||
| Time1 | 26.6 (2.6) | 25.9 (3.4) | 28.2 (2.5) | 29.5 (0.6)h | <0.001 | |||||
| Time2 | 26.2 (3.1) | 26.4 (3.4) | 27.8 (2.3) | |||||||
| FAB | ||||||||||
| Time1 | 12.6 (2.8) | 14.6 (2.8) | 17.0 (1.1)i | – | <0.001 | |||||
| Time2 | 10.5 (4.4) | 13.2 (3.3) | 16.1 (2.4)j | – | 0.003 | |||||
Post hoc analyses: aPD versus PSP/MSA-P, P < 0.001; bPD versus MSA-P, P = 0.002; cPD versus MSA-P, P = 0.004, PD versus PSP, P = 0.01; dPD versus MSA-P, P = 0.04; ePD versus MSA-P, P = 0.03; fPD versus PSP, P = 0.05, PD versus MSA-P, P = 0.003; gPD versus PSP, P = 0.007, PD versus MSA-P, P = 0.005; hcontrol versus PSP, P = 0.004, control versus MSA-P, P = 0.003; iPD versus PSP, P < 0.001; jPD versus PSP, P = 0.003; PD = Parkinson's disease.
Levodopa responsiveness in the PSP and MSA-P patients was as follows: PSP, no trial, four; no response, nine; moderate response, three; good response, one; MSA-P, no trial, two; no response, one; moderate response, three; good response, two; excellent response, one.
Intraclass correlation coefficients for midbrain, pons, cerebellum and third ventricle volume measurements were all greater than 0.9, suggesting a high level of reliability.
The results of cross sectional regional volume measurements in these patients confirmed that midbrain volume is reduced in PSP and pons and cerebellar volumes are lower in MSA-P (Paviour et al., 2005a).
Using the BSI to calculate whole brain and regional atrophy rates, rather than the difference in manually measured volumes, significantly reduced the variability in the atrophy rates reported for whole brain (P < 0.001), midbrain (P = 0.001), pons (P = 0.001) total frontal (P = 0.002) and PI (P < 0.001) regions, but not for the SCP, the lateral or third ventricles.
BSI derived annualized rates of atrophy (% year−1) are reported in Table 2 and show that regional rates of brainstem atrophy are greater than whole-brain rates of atrophy in PSP and MSA-P. This was confirmed by comparing regional rates of atrophy in PSP and MSA-P with the whole brain rate of atrophy using a two sample t-test. In PSP, midbrain and SCP atrophy rates were greater than whole brain atrophy rates (P < 0.001 and P = 0.02). In MSA-P, pontine and cerebellar atrophy rates were greater than whole brain atrophy rates (P = 0.004 and P = 0.002).
Mean (SD) percentage tissue lost per annum
| PSP | MSA-P | PD | Controls | ||
|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ANOVA significance levels |
| Brain | 1.2 (1.0)a | 1.0 (1.1) | 0.6 (0.7) | 0.4 (0.5) | 0.04 |
| Frontal | 1.2 (1.3) | 0.9 (1.2) | 0.6 (0.8) | 0.4 (0.5) | 0.13 |
| PI | 1.0 (1.0) | 1.4 (1.2)b | 0.8 (0.6) | 0.3 (0.5) | 0.008 |
| Mann–Whitney U significance levels | |||||
| Midbrain | 2.2 (1.5) | 1.8 (1.9) | 0.6 (0.9) | 0.3 (0.6) | PSP versus PD P = 0.004, versus controls P < 0.001; MSA versus controls P = 0.006 |
| Pons | 1.5 (1.3) | 4.5 (3.2) | 0.4 (0.5) | 0.2 (0.3) | MSA versus PSP P = 0.03, versus PD P = 0.002; MSA/PSP versus controls P < 0.001; PSP versus PD P = 0.01 |
| Cerebellum | 1.4 (1.1) | 3.2 (1.9) | 0.8 (0.6) | 0.3 (1.0) | MSA versus PSP P = 0.02, versus PD P = 0.005, versus controls P < 0.001; PSP versus controls P = 0.009 |
| SCP | 3.5 (4.0) | 3.7 (10.0) | −1.1 (2.0) | 0 (2.5) | PSP versus PD P = 0.004, versus controls P = 0.008; MSA versus PD P = 0.05, versus controls P = 0.03 |
| Third ventricle | 6.1 (6.0) | 7.6 (10.1) | 4.7 (3.9) | 2.2 (3.8) | PSP versus controls P = 0.02 |
| Lateral ventricle | 9.6 (9.8) | 9.5 (12.4) | 5.3 (5.2) | 3.3 (3.3) | PSP versus controls P = 0.008 |
| PSP | MSA-P | PD | Controls | ||
|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ANOVA significance levels |
| Brain | 1.2 (1.0)a | 1.0 (1.1) | 0.6 (0.7) | 0.4 (0.5) | 0.04 |
| Frontal | 1.2 (1.3) | 0.9 (1.2) | 0.6 (0.8) | 0.4 (0.5) | 0.13 |
| PI | 1.0 (1.0) | 1.4 (1.2)b | 0.8 (0.6) | 0.3 (0.5) | 0.008 |
| Mann–Whitney U significance levels | |||||
| Midbrain | 2.2 (1.5) | 1.8 (1.9) | 0.6 (0.9) | 0.3 (0.6) | PSP versus PD P = 0.004, versus controls P < 0.001; MSA versus controls P = 0.006 |
| Pons | 1.5 (1.3) | 4.5 (3.2) | 0.4 (0.5) | 0.2 (0.3) | MSA versus PSP P = 0.03, versus PD P = 0.002; MSA/PSP versus controls P < 0.001; PSP versus PD P = 0.01 |
| Cerebellum | 1.4 (1.1) | 3.2 (1.9) | 0.8 (0.6) | 0.3 (1.0) | MSA versus PSP P = 0.02, versus PD P = 0.005, versus controls P < 0.001; PSP versus controls P = 0.009 |
| SCP | 3.5 (4.0) | 3.7 (10.0) | −1.1 (2.0) | 0 (2.5) | PSP versus PD P = 0.004, versus controls P = 0.008; MSA versus PD P = 0.05, versus controls P = 0.03 |
| Third ventricle | 6.1 (6.0) | 7.6 (10.1) | 4.7 (3.9) | 2.2 (3.8) | PSP versus controls P = 0.02 |
| Lateral ventricle | 9.6 (9.8) | 9.5 (12.4) | 5.3 (5.2) | 3.3 (3.3) | PSP versus controls P = 0.008 |
ANOVA Post hoc analyses: acontrols versus PSP, P = 0.045; bMSA versus controls, P = 0.008. PI-posterior inferior region, SCP-superior cerebellar peduncle.
Mean (SD) percentage tissue lost per annum
| PSP | MSA-P | PD | Controls | ||
|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ANOVA significance levels |
| Brain | 1.2 (1.0)a | 1.0 (1.1) | 0.6 (0.7) | 0.4 (0.5) | 0.04 |
| Frontal | 1.2 (1.3) | 0.9 (1.2) | 0.6 (0.8) | 0.4 (0.5) | 0.13 |
| PI | 1.0 (1.0) | 1.4 (1.2)b | 0.8 (0.6) | 0.3 (0.5) | 0.008 |
| Mann–Whitney U significance levels | |||||
| Midbrain | 2.2 (1.5) | 1.8 (1.9) | 0.6 (0.9) | 0.3 (0.6) | PSP versus PD P = 0.004, versus controls P < 0.001; MSA versus controls P = 0.006 |
| Pons | 1.5 (1.3) | 4.5 (3.2) | 0.4 (0.5) | 0.2 (0.3) | MSA versus PSP P = 0.03, versus PD P = 0.002; MSA/PSP versus controls P < 0.001; PSP versus PD P = 0.01 |
| Cerebellum | 1.4 (1.1) | 3.2 (1.9) | 0.8 (0.6) | 0.3 (1.0) | MSA versus PSP P = 0.02, versus PD P = 0.005, versus controls P < 0.001; PSP versus controls P = 0.009 |
| SCP | 3.5 (4.0) | 3.7 (10.0) | −1.1 (2.0) | 0 (2.5) | PSP versus PD P = 0.004, versus controls P = 0.008; MSA versus PD P = 0.05, versus controls P = 0.03 |
| Third ventricle | 6.1 (6.0) | 7.6 (10.1) | 4.7 (3.9) | 2.2 (3.8) | PSP versus controls P = 0.02 |
| Lateral ventricle | 9.6 (9.8) | 9.5 (12.4) | 5.3 (5.2) | 3.3 (3.3) | PSP versus controls P = 0.008 |
| PSP | MSA-P | PD | Controls | ||
|---|---|---|---|---|---|
| n | 17 | 9 | 9 | 18 | ANOVA significance levels |
| Brain | 1.2 (1.0)a | 1.0 (1.1) | 0.6 (0.7) | 0.4 (0.5) | 0.04 |
| Frontal | 1.2 (1.3) | 0.9 (1.2) | 0.6 (0.8) | 0.4 (0.5) | 0.13 |
| PI | 1.0 (1.0) | 1.4 (1.2)b | 0.8 (0.6) | 0.3 (0.5) | 0.008 |
| Mann–Whitney U significance levels | |||||
| Midbrain | 2.2 (1.5) | 1.8 (1.9) | 0.6 (0.9) | 0.3 (0.6) | PSP versus PD P = 0.004, versus controls P < 0.001; MSA versus controls P = 0.006 |
| Pons | 1.5 (1.3) | 4.5 (3.2) | 0.4 (0.5) | 0.2 (0.3) | MSA versus PSP P = 0.03, versus PD P = 0.002; MSA/PSP versus controls P < 0.001; PSP versus PD P = 0.01 |
| Cerebellum | 1.4 (1.1) | 3.2 (1.9) | 0.8 (0.6) | 0.3 (1.0) | MSA versus PSP P = 0.02, versus PD P = 0.005, versus controls P < 0.001; PSP versus controls P = 0.009 |
| SCP | 3.5 (4.0) | 3.7 (10.0) | −1.1 (2.0) | 0 (2.5) | PSP versus PD P = 0.004, versus controls P = 0.008; MSA versus PD P = 0.05, versus controls P = 0.03 |
| Third ventricle | 6.1 (6.0) | 7.6 (10.1) | 4.7 (3.9) | 2.2 (3.8) | PSP versus controls P = 0.02 |
| Lateral ventricle | 9.6 (9.8) | 9.5 (12.4) | 5.3 (5.2) | 3.3 (3.3) | PSP versus controls P = 0.008 |
ANOVA Post hoc analyses: acontrols versus PSP, P = 0.045; bMSA versus controls, P = 0.008. PI-posterior inferior region, SCP-superior cerebellar peduncle.
For all regional atrophy rates which discriminated the patient groups, logistic regression analysis was applied to assess whether atrophy rates were better than cross sectional volumes at discriminating PSP or MSA-P from each other as well as from Parkinson's disease patients and healthy controls. Only cerebellar atrophy rates were better discriminators of MSA-P from PSP, Parkinson's disease patients and healthy controls (P = 0.01)
While rates of BSI derived pontine atrophy (P = 0.02) were marginally better than cross sectional total intracranial volume corrected pontine volume (P = 0.04) at discriminating between MSA-P and PSP, logistic regression analysis suggested this was not significant (P = 0.08). BSI derived rates of cerebellar atrophy were significantly better than cross sectional total intracranial volume corrected cerebellar volume at discriminating between MSA-P and PSP (P = 0.04).
Clinico-radiological correlations
PSP
An increased rate of BSI derived midbrain atrophy in PSP was associated with: a decline in the Mattis DRS-attention sub-test (Corr. = 0.80, P = 0.009); an increasing number of errors (but not perseverative errors) on the WCST (Corr. = 0.72, P = 0.04); a decrease in the PASAT score (Corr. = 0.89, P = 0.04); a decrease in FAB score (Corr. = 0.70, P = 0.008); an increase in motor disability (UPDRSIII) score (Corr. = 0.58, P = 0.04) and a decrease in the MMSE (Corr. = 0.56, P = 0.04). Increased pontine atrophy rates were only associated with a decrease in FAB score (P < 0.05). Increasing cerebellar atrophy rates were associated with a decrease in FAB score (Corr. = 0.73, P = 0.005) and in the MMSE (Corr. = 0.55, P = 0.05).
Increased whole brain atrophy rates were associated with decline in performance on: the Mattis DRS attention sub-test (Corr. = 0.77, P = 0.009); an increasing number of errors on the WCST (Corr. = 0.83, P = 0.006); an increase in the number of intrusions on the verbal fluency test (Corr. = 0.89, P = 0.003); a decline in performance on the PASAT (Corr. = 0.82, P = 0.045); and a reducing score on the FAB (Corr. = 0.65, P = 0.02) and the MMSE (Corr. = 0.68, P = 0.01).
Increased rates of frontal and postero-inferior atrophy were similarly associated with a more severe deterioration in: the Mattis DRS attention sub-test (frontal, Corr. = 0.70, P = 0.02; postero-inferior, Corr. = 0.65, P = 0.04); the number of errors on the WCST (frontal, Corr. = 0.85, P = 0.004; postero-inferior, Corr. = 0.72, P = 0.03); a decline in the PASAT score (frontal, Corr. = 0.94, P = 0.005; postero-inferior, Corr. = 0.83, P = 0.04); the FAB (frontal, Corr. = 0.62, P = 0.02; postero-inferior, Corr. = 0.67, P = 0.01); the MMSE (frontal, Corr. = 0.56, P = 0.05; postero-inferior, Corr. = 0.75, P = 0.003). An increasing rate of postero-inferior atrophy was also associated with increased motor disability measured on the UPDRSIII (Corr. = 0.56, P = 0.05).
An increasing rate of third ventricle enlargement was associated with a decline in performance on: the DRS attention subtest (Corr. = 0.73, P = 0.02,); the number of errors on the WCST (Corr. = 0.80, P = 0.009); an increasing number of intrusions on the verbal fluency test (Corr. = 0.81, P = 0.014) and a decline on the PASAT (Corr. = 0.93, P = 0.008). An increasing rate of lateral ventricle enlargement was only associated with a decline in the FAB (P < 0.05) and a decline in the MMSE (P < 0.05).
Having adjusted for age and disease duration, decline in the FAB score was most strongly associated with midbrain atrophy rate (P = 0.008). The next strongest association was with atrophy rate in the cerebellum (P = 0.005), but neither of these had independent effects once the other was controlled for. Change in UPDRSIII was most strongly associated with midbrain atrophy (P = 0.04) and then posterior inferior atrophy. Again, neither of these had independent effects once the other was controlled for. Similar results were obtained for change in the MMSE.
MSA-P
In the MSA-P patients, increased rates of pontine atrophy were associated with a decline in performance on: the DRS total score (Corr. = 0.99, P = 0.001); the digit span (Corr. = 0.88, P = 0.05) and semantic verbal fluency (Corr. = 0.95, P = 0.05) as well as an increase in the motor disability as scored on the UPDRSIII (Corr. = 0.8, P = 0.05).
Increasing cerebellar atrophy rates were also associated with declining performance on the DRS total score (Corr. = 0.96, P = 0.01); the DRS conceptualization subtest (Corr. = 0.89, P = 0.04); semantic verbal fluency (Corr. = 0.95, P = 0.05) and UPDRSIII (Corr. = 0.86, P = 0.03).
Increasing midbrain atrophy rates were associated with poorer performance on: the DRS initiation and perseveration subtest (Corr. = 0.97, P = 0.005); the recognition memory test for faces (Corr.= 0.92, P = 0.03); the digit span (Corr. = 0.91, P = 0.03) and intrusions on the verbal fluency test (Corr. = 0.96, P = 0.04).
Few other associations were noted in the MSA-P group although lower whole brain, frontal and postero-inferior atrophy rates were associated with a greater rate of decline in the PASAT score (P < 0.05) and for frontal atrophy rates, the Beck Anxiety Inventory (P = 0.04).
In the control subjects, an association between age and increasing rates of lateral ventricle enlargement (P = 0.04) and posterior-inferior atrophy (P = 0.04) were noted. No associations between age or disease duration and atrophy rates were seen in PSP or MSA-P.
As with the associations noted in the PSP cohort, multiple regression analysis confirmed that change in motor deficit was most strongly associated with cerebellar and pontine atrophy rates but that neither of these had independent effects once the other was controlled for.
Discussion
This study is the first to quantify regional as well as whole brain atrophy rates during life in PSP, MSA-P and Parkinson's disease based on registration of serial volumetric MRI brain scans. We demonstrate that regional rates of brainstem atrophy are greater than whole-brain rates of atrophy in PSP and MSA-P. These regional atrophy rates differentiate PSP and MSA-P from each other and from Parkinson's disease patients and healthy controls, whereas whole brain atrophy rates are only able to distinguish PSP from healthy control subjects, suggesting regional rather than whole brain atrophy rates may have more clinical utility. Mean rates of midbrain atrophy in PSP were seven times the mean rate in controls. SCP atrophy rates were greatest in PSP, but the variability in the measured atrophy rate limited the significance of the group differences. Pontine atrophy rates were greatest in MSA-P (20 times the mean rate in controls and three times the mean rate in PSP). Atrophy rates in the cerebellum discriminate MSA-P from PSP with greater accuracy than the corresponding regional volume measured at a single time point.
Using the BSI derived, rather than manually calculated measurements reduced the variability seen in regional atrophy rates. Differences between the mean BSI derived and manually derived atrophy rates are likely to arise because the BSI measurements are less prone to operator error.
The rates of whole brain atrophy measured in our study are of the same magnitude as those recently published in a smaller cohort of PSP patients (Josephs et al., 2006) with a later disease onset (72.8 years) and a longer mean scan interval (3.1 years) than in our study. It is important that our study confirms brain atrophy rates are similar in PSP patients with a more typical age of onset and that using a shorter interval between scans is still practical in terms of atrophy rate calculations. One of the PSP patients studied by Josephs et al. had an initial scan two years prior to disease onset. The rate of brain atrophy in the first two asymptomatic years was 0.9% per annum suggesting that accelerated atrophy rates may precede symptoms. An increasing rate of regional atrophy may occur with a similar temporal relationship to clinical symptoms and if so, the results of our study suggest that regional rather than whole brain atrophy rates would better identify patients in the earliest stages of the disease when potential disease modifying therapies would be most effective.
Establishing the rate of whole brain and regional atrophy in a larger group of PSP patients with detailed, prospective collection of clinical information as we have done is important. The disease is heterogeneous and relying on a small number of PSP subjects, even with pathological confirmation (Josephs et al., 2006), is not ideal. There are a number of associations between atrophy rates and the clinical features of these diseases. Crucially, rates of atrophy in PSP and MSA-P have clinical correlates reflecting progression in clinically meaningful end-points. This is important if atrophy rates are to be applied as a measure of progression in a trial of a disease modifying therapy and as such this study helps to evaluate the use of atrophy rates in this context. The most significant examples are increasing midbrain atrophy rates and worsening motor deficit in PSP, but not MSA-P. Conversely, worsening motor deficit was associated with increased ponto-cerebellar atrophy in MSA-P but not PSP. This suggests that pathological involvement of the structures in these regions, represented by tissue loss (and presumably cell death), is strongly associated with progression in the critical clinical features of the diseases. Hence regional MRI derived atrophy rates, which correlate with change in motor deficit in PSP and MSA-P may be useful and quantifiable outcome measures in clinical trials.
Associations between atrophy rates and change in neuropsychological and clinical scores were largely confined to subtests of the Mattis DRS, the WCST and the PASAT. Associations between increasing rate of frontal atrophy in PSP and an increasing number of errors on the WCST, and declining PASAT and FAB scores, suggest that the degree of severity noted in the dysexecutive syndrome in PSP is related to frontal atrophy. This supports observations from cross sectional imaging studies (Cordato et al., 2002; Cordato et al., 2005). Recent studies have identified localized areas of frontal atrophy such as the cingulate gyrus and medial frontal gyrus (Cordato et al., 2002; Brenneis et al., 2004). Atrophy rates in these specific regions may be more strongly associated with executive dysfunction, but segmentation of these regions would be time consuming and dependent on arbitrary decisions and subjective judgements in outlining, and have not been undertaken. Damage to brainstem and sub-cortical structures also contributes to the cognitive dysfunction in PSP and hence it is not surprising that there is an association between a decline in the FAB score and increased rates of midbrain atrophy in PSP and in fact the association with midbrain atrophy was stronger than with frontal atrophy.
The association between a greater decline in the FAB score in PSP and increasing lateral ventricle atrophy rates could in part be explained by changes at the lateral ventricle/caudate nucleus boundary. Atrophy of the caudate nucleus in PSP would contribute to a worsening sub-cortical dementia, which might help to explain the clinical association. However the caudate nucleus makes up only a small part of the lateral ventricle CSF/brain tissue boundary. Hence it is probable that lateral ventricle volume changes do not reflect tissue loss from immediately around the structure and are more likely to arise as a consequence of distant tissue loss and subsequent structural readjustments. Tissue loss in the frontal lobes could contribute to ventricular enlargement in this way and could help to explain the associations detected.
Correlations were also seen between some of these neuropsychological tests and whole brain as well as posterior inferior atrophy rates in the PSP patients. It is possible that the associations arise because the tests applied are the most sensitive to change over a relatively short period of time. Other tests may be less sensitive or may be at floor at the first assessment meaning no associations would be detected.
A large number of regression analyses were performed to assess the relationship between regional atrophy rates and disease progression. The results of multiple regression models, using age, disease duration and the next most significantly associated regional atrophy rate as covariates suggest that some of the associations seen between regional atrophy rates and change in disease severity arise because atrophy in one region is likely to occur in parallel with another region. For example, the association between pontine atrophy and change in the FAB score in PSP may only arise because the real association is with midbrain atrophy, and pontine atrophy and midbrain atrophy occur in parallel. Including the next most significantly associated regional atrophy rate as a covariate helps to overcome some of these problems. Similarly, the smaller number of patients with MSA-P may have limited the associations detected in this group, even though all were of the parkinsonian phenotype. Larger study numbers would help to overcome all of these limitations and clarify where true associations occur.
Notably, ponto-cerebellar atrophy rates are increased in PSP compared to healthy controls and midbrain atrophy rates are increased in MSA-P compared to controls. In PSP, pathological involvement of the deep cerebellar nuclei and the pons is well established (Daniel et al., 1995). The cerebellar cortex may also be involved (Piao et al., 2002), explaining the ponto-cerebellar atrophy observed. The localized pattern of atrophy in the cerebellum and the pons in PSP and MSA-P is likely to be different. In MSA-P, atrophy of the cerebellar cortex and vermis is likely to be more severe than in PSP and midbrain atrophy (which may arise secondary to pathology in the substantia nigra) less severe than in PSP as pathological involvement of the SCP, red nucleus and the superior colliculus would also contribute to the increased atrophy rates in PSP.
It is likely that pathological involvement of basal ganglia regions such as the globus pallidus, putamen and caudate nucleus also contribute to the clinical features of these disorders. Tissue boundaries in these regions may be less distinct and hence MRI measurements of atrophy less reliable and more labour intensive. This would limit the utility of basal ganglia atrophy rates as a marker of disease progression, as few clinico-radiological associations may be detected, even if associations with the clinical features of the disease could be readily explained.
Serial volumetric imaging studies have largely been confined to single case reports or individual cases within the context of cross sectional studies (Schott et al., 2003; Paviour et al., 2004). A previous large study of MRI changes over time in MSA relied largely on qualitative imaging changes, but provided useful information regarding the development of certain imaging features over time (Horimoto et al., 2002). Other studies have utilised area measurements to determine atrophy rates in MSA (Horimoto et al., 2000; Konagaya et al., 2002; Watanabe et al., 2002). Konagaya et al. studied nine MSA patients in whom the mean scan interval was 7 years. The ratio of cerebral hemisphere area to intracranial area decreased from a mean of 82–70% suggesting an atrophy rate of 2% per annum. Atrophy rates reported in our study are of a lower magnitude. However area measurements cannot be directly extrapolated to volume change due to the non-uniform distribution of brain tissue loss, as demonstrated in this study. As such, whole brain atrophy rates based on area measurements may give different rates depending upon at which level the area is measured, although the true rate of whole brain atrophy is the same. In fact, longitudinal imaging studies based on area measurements in MSA support non-uniform atrophy, and conclude that cerebellar and pontine atrophy are more significant in MSA with predominant cerebellar signs (Watanabe et al., 2002). The pons and cerebellum had the greatest rates of atrophy in the MSA-P patients in our study. No rating scale assessing cerebellar function was undertaken. However, while the dominant clinical phenotype was of MSA-P, five of the MSA-P patients fulfilled the criterion for cerebellar dysfunction according to the clinical diagnostic criteria for MSA (Gilman et al., 1999). The patient with pathological confirmation of MSA had OPCA predominant pathology and despite having mild cerebellar signs, had predominant parkinsonism. In a large series of pathologically proven cases, 40% of OPCA predominant cases did not have cerebellar signs documented during life. This confirms that the frequency of cerebellar signs in MSA is low even in cases with obvious pathological involvement of the cerebellum and that atrophy in the cerebellum and the pons is still significant when Parkinsonism is the predominant clinical feature as the degree of striatonigral pathology required for a Parkinsonian syndrome is relatively less than the olivo-ponto-cerebellar pathology required for a predominant cerebellar phenotype (Ozawa et al., 2004).
The gold standard method for assessing regional volumes remains manual outlining of the structure of interest. The BSI is semi automated and because it calculates change directly from differences between MR images, is less vulnerable to errors of manual outlining. It has also been shown to be a repeatable and reliable measure of whole brain atrophy (Fox and Freeborough, 1997). The advantage of using the local BSI is that it is possible to assess a small region known to be susceptible to pathological involvement rather than the whole brain, which in PSP and MSA-P includes regions that may not be affected by the pathological process. The results of this study confirm that in these neurodegenerative diseases, in which the pathological load has a predilection for brainstem structures, regional rather than whole brain atrophy rates are better markers of disease progression.
The rates of regional atrophy within the subject groups reported in this study are heterogeneous. This can partly be explained by manual segmentation errors or scan artefact, but a significant component is likely to be due to true differences in rates of disease progression between individual patients and because individual atrophy rates may vary during the course of the disease. These factors may also underlie the lack of significant associations between disease duration and atrophy rates. This may limit the utility of serial MRI in evaluating disease progression in natural history studies. Alternatively, it may be that there is truly no association between disease duration and atrophy rates. One study based on area measurements, found only weak associa-tions between rates of atrophy and measures of disease severity suggesting that atrophy rates may be relatively constant throughout the disease (Konagaya et al., 2002).
A limitation of this study is that only three patients have had the clinical diagnosis confirmed at post mortem. However, the remaining patients have been followed up in a tertiary centre where the clinical diagnostic accuracy is well established (Hughes et al., 2002).
Image analysis methods cannot completely control for differences in scan acquisition and errors may arise due to registration difficulty because of movement artefact or quality differences between the scans. Inevitably, with all of these regional measurements, there are arbitrary decisions about which tissue to include. Errors related to these measurements as well as acquisition may occur and important changes may in fact be under-reported.
The BSI is likely to under report change compared with simple subtraction of repeat from baseline volumes. In addition, it does not eradicate all error in measuring the region concerned. Even when using the BSI, negative values of tissue loss occur, suggesting erroneous growth. Minor registration errors or quality differences between baseline and registered repeat images may contribute to this. These problems still occur when the volume loss is calculated by subtracting the repeat image volume from the baseline volume.
Atrophy rates in these diseases may well be greater than in healthy controls even in the earliest stages of the illness, but this remains to be tested. Early MRI studies with a repeat scan in 6–9 months may be warranted when symptoms and signs are subtle, in order to identify a neurodegenerative disease process. It is also in the very early stages that a disease-modifying drug may be at its most useful and so early identification is likely to become increasingly important.
In summary, regional rates of atrophy clearly distinguish PSP and MSA-P from Parkinson's disease patients and healthy controls as well as from each other. Rates of whole brain atrophy derived from registered serial MRI brain scans are greater in PSP than in controls. The measured rates of regional atrophy correlate with clinical measures of disease progression, particularly with regard to motor disability scales in PSP and MSA-P. BSI derived rates of atrophy reduce variability compared to manually derived atrophy rates and these conclusions may be important if atrophy rates are to be used as outcome measures in clinical trials.
D.C.P is supported by a grant from the PSP (Europe) Association, N.C.F holds a Medical Research Council Senior Clinical Fellowship. Chris Frost MA, DipStat (Department of Epidemiology and Population Health, Medical Statistics Unit, London School of Hygiene and Tropical Medicine, London, UK) advised on statistical methodology.
References
Asato R, Akiguchi I, Masunaga S, Hashimoto N. Magnetic resonance imaging distinguishes progressive supranuclear palsy from multiple system atrophy.
Barnes J, Scahill RI, Boyes RG, Frost C, Lewis EB, Rossor CL, et al. Differentiating AD from aging using semiautomated measurement of hippocampal atrophy rates.
Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression.
Benton AL. Differential behavioral effects in frontal lobe disease.
Brenneis C, Seppi K, Schocke M, Benke T, Wenning GK, Poewe W. Voxel based morphometry reveals a distinct pattern of frontal atrophy in progressive supranuclear palsy.
Brenneis C, Seppi K, Schocke MF, Muller J, Luginger E, Bosch S, et al. Voxel-based morphometry detects cortical atrophy in the Parkinson variant of multiple system atrophy.
Chan D, Fox NC, Jenkins R, Scahill RI, Crum WR, Rossor MN. Rates of global and regional cerebral atrophy in AD and frontotemporal dementia.
Chan D, Fox NC, Scahill RI, Crum WR, Whitwell JL, Leschziner G, et al. Patterns of temporal lobe atrophy in semantic dementia and Alzheimer's disease.
Cordato NJ, Pantelis C, Halliday GM, Velakoulis D, Wood SJ, Stuart GW, et al. Frontal atrophy correlates with behavioural changes in progressive supranuclear palsy.
Cordato NJ, Duggins AJ, Halliday GM, Morris JGL, Pantelis C. Clinical deficits correlate with regional cerebral atrophy in progressive supranuclear palsy.
Dalton CM, Brex PA, Jenkins R, Fox NC, Miszkiel KA, Crum WR, et al. Progressive ventricular enlargement in patients with clinically isolated syndromes is associated with the early development of multiple sclerosis.
Daniel SE, de Bruin VM, Lees AJ. The clinical and pathological spectrum of Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy): a reappraisal.
Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a Frontal Assessment Battery at bedside.
Fahn S, Elton R. Members of the UPDRS development committee. Recent developments in Parkinson's disease, Vol 2. Florham Park, NJ: Macmillan Health Care Information;
Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician.
Fox NC, Freeborough PA. Brain atrophy progression measured from registered serial MRI: validation and application to Alzheimer's disease.
Fox NC, Freeborough PA, Rossor MN. Visualisation and quantification of rates of atrophy in Alzheimer's disease.
Fox NC, Scahill RI, Crum WR, Rossor MN. Correlation between rates of brain atrophy and cognitive decline in AD.
Freeborough PA, Fox NC. The boundary shift integral: an accurate and robust measure of cerebral volume changes from registered repeat MRI.
Freeborough PA, Fox NC, Kitney RI. Interactive algorithms for the segmentation and quantitation of 3-D MRI brain scans.
Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease.
Gilman S, Low PA, Quinn N, Albanese A, Ben Shlomo Y, Fowler CJ, et al. Consensus statement on the diagnosis of multiple system atrophy.
Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy.
Gronwall D, Wrightson P. Memory and information processing capacity after closed head injury.
Groschel K, Hauser TK, Luft A, Patronas N, Dichgans J, Litvan I, et al. Magnetic resonance imaging-based volumetry differentiates progressive supranuclear palsy from corticobasal degeneration.
Horimoto Y, Aiba I, Yasuda T, Ohkawa Y, Katayama T, Yokokawa Y, et al. Cerebral atrophy in multiple system atrophy by MRI.
Horimoto Y, Aiba I, Yasuda T, Ohkawa Y, Katayama T, Yokokawa Y, et al. Longitudinal MRI study of multiple system atrophy—when do the findings appear, and what is the course?
Hughes AJ, Daniel SE, Ben Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service.
Jack CR Jr, Petersen RC, Xu Y, O'Brien PC, Smith GE, Ivnik RJ, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD.
Josephs KA, Whitwell JL, Boeve BF, Shiung MM, Gunter JL, Parisi JE, et al. Rates of cerebral atrophy in autopsy-confirmed progressive supranuclear palsy.
Konagaya M, Konagaya Y, Sakai M, Matsuoka Y, Hashizume Y. Progressive cerebral atrophy in multiple system atrophy.
Lewis EB, Fox NC. Correction of differential intensity inhomogeneity in longitudinal MR images.
Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I, et al. SIC Task Force appraisal of clinical diagnostic criteria for parkinsonian disorders.
Mattis S. Mental status examination for organic mental syndrome in the elderly patients. In: Bellack L, Karasu TB, editors. Geriatric psychiatry. New York: Grune & Stratton;
Nath U, Ben Shlomo Y, Thomson RG, Morris HR, Wood NW, Lees AJ, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK.
Oba H, Yagishita A, Terada H, Barkovich AJ, Kutomi K, Yamauchi T, et al. New and reliable MRI diagnosis for progressive supranuclear palsy.
Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations.
Paviour DC, Schott JM, Stevens JM, Revesz T, Holton JL, Rossor MN, et al. Pathological substrate for regional distribution of increased atrophy rates in progressive supranuclear palsy.
Paviour D, Price SL, Jahanshahi M, Lees AJ, Fox NC. Regional brain volumes distinguish PSP, MSA-P and PD: MRI based clinico-radiological correlations.
Paviour DC, Price SL, Stevens JM, Lees AJ, Fox NC. Quantitative MRI measurement of superior cerebellar peduncle in progressive supranuclear palsy.
Paviour DC, Winterburn D, Simmonds S, Burgess G, Wilkinson L, Fox NC, et al. Can the frontal assessment battery (FAB) differentiate bradykinetic rigid syndromes? Relation of the FAB to formal neuropsychological testing.
Piao YS, Hayashi S, Wakabayashi K, Kakita A, Aida I, Yamada M, et al. Cerebellar cortical tau pathology in progressive supranuclear palsy and corticobasal degeneration.
Price S, Paviour DC, Scahill R, Stevens J, Rossor MN, Lees AJ. Voxel-based morphometry detects patterns of atrophy that help differentiate progressive supranuclear palsy and Parkinson's disease.
Reitan RM. Validity of the trail making test as an indicator of organic brain damage.
Schott JM, Simon JE, Fox NC, King AP, Khan MN, Cipolotti L, et al. Delineating the sites and progression of in vivo atrophy in multiple system atrophy using fluid-registered MRI.
Schrag A, Good CD, Miszkiel K, Morris HR, Mathias CJ, Lees AJ, et al. Differentiation of atypical parkinsonian syndromes with routine MRI.
Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy.
Stern MB, Braffman BH, Skolnick BE, Hurtig HI, Grossman RI. Magnetic resonance imaging in Parkinson's disease and parkinsonian syndromes.
Warrington EK. Manual for the Recognition Memory Test for words and faces. Windsor: NFER-NELSON;
Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients.
Weschler D. Manual for the Wechsler Adult Intelligence Scale-Revised. New York: Psychological Corp;
Whitwell JL, Crum WR, Watt HC, Fox NC. Normalization of cerebral volumes by use of intracranial volume: implications for longitudinal quantitative MR imaging.
Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP-parkinsonism.
Author notes
1The Sara Koe PSP Research Centre, Institute of Neurology UCL, 2Dementia Research Centre (UCL), Institute of Neurology and 3The Cognitive-Motor Neuroscience Group, Sobell Department of Motor Neuroscience and Movement Disorders, Institute of Neurology, Queen Square, 4Rita Lila Weston Institute of Neurological Studies, UCL, London, UK


{kind=link}
{kind=link}
{kind=link}