
Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review

 , , ,
, , ,
Abstract
:
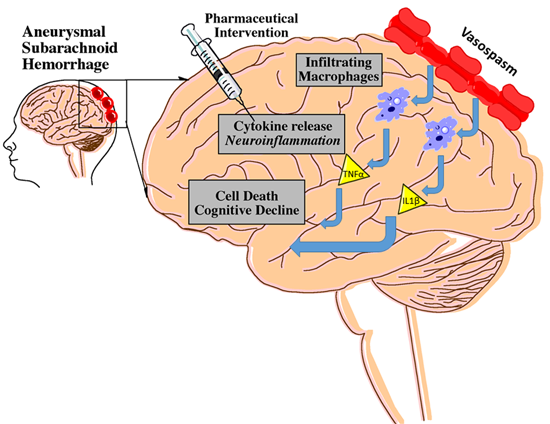
1. Introduction
2. Neuroinflammation
2.1. The Role of Inflammation
2.2. SAH Pathophysiology: Acute Events
2.3. SAH Pathophysiology: Subacute-Chronic Events
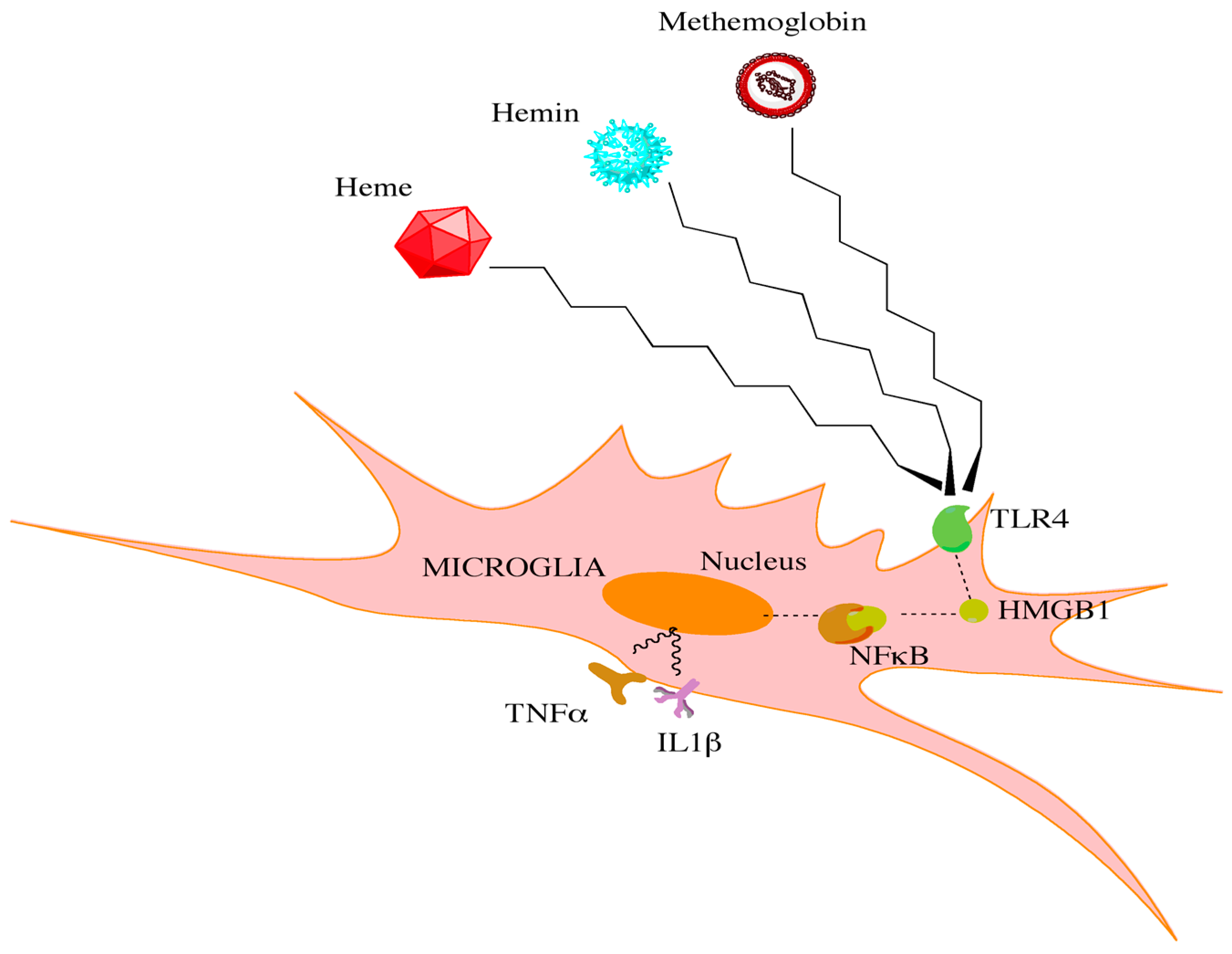
2.4. Inflammatory Mediators in SAH: A Focus on Cytokines and Cell Lines
2.5. Inflammatory Mediators in SAH: A Focus on Proteases
2.6. SAH-Associated Inflammation: An Inflow or an Outflow Problem?
3. Secondary Outcomes
3.1. Cerebral Vasospasm
3.2. CV and Inflammation
3.3. CV and Long-Term Deficits
4. Etiology and Comorbidities
4.1. Genetic Factors
4.2. Comorbidities and Physical Correlates
5. Targeting Neuroinflammation
5.1. Treatment
5.2. Lessons from Animal Models
5.3. Novel Discoveries
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, B.A.; Turan, N.; Chau, M.; Pradilla, G. Inflammation, vasospasm, and brain injury after subarachnoid hemorrhage. BioMed Res. Int. 2014, 2014, 384342. [Google Scholar] [CrossRef] [PubMed]
- Guresir, E.; Vasiliadis, N.; Konczalla, J.; Raab, P.; Hattingen, E.; Seifert, V.; Vatter, H. Erythropoietin prevents delayed hemodynamic dysfunction after subarachnoid hemorrhage in a randomized controlled experimental setting. J. Neurol. Sci. 2013, 332, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Caffes, N.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Glibenclamide for the treatment of ischemic and hemorrhagic stroke. Int. J. Mol. Sci. 2015, 16, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Tada, Y.; Wada, K.; Liang, E.I.; Chang, M.; Mobashery, S.; Kanematsu, Y.; Kurihara, C.; Palova, E.; Kanematsu, M.; et al. Pharmacological stabilization of intracranial aneurysms in mice: A feasibility study. Stroke 2012, 43, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.; Grille, S.; Morelli, P.; Mila, R.; Trias, N.; Brugnini, A.; N, L.L.; Biestro, A.; Lens, D. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with aneurysmal subarachnoid hemorrhage. SpringerPlus 2015, 4, 195. [Google Scholar] [CrossRef] [PubMed]
- Kooijman, E.; Nijboer, C.H.; van Velthoven, C.T.; Kavelaars, A.; Kesecioglu, J.; Heijnen, C.J. The rodent endovascular puncture model of subarachnoid hemorrhage: Mechanisms of brain damage and therapeutic strategies. J. Neuroinflamm. 2014, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Badjatia, N. Participants in the International Multi-disciplinary Consensus Conference on Multimodality, M. Monitoring inflammation (including fever) in acute brain injury. Neurocrit. Care 2014, 21, S177–S186. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Garcia, M.; Testai, F.; Vetri, F.; Barabanova, A.; Pelligrino, D.A.; Paisansathan, C. Pharmacologic blockade of vascular adhesion protein-1 lessens neurologic dysfunction in rats subjected to subarachnoid hemorrhage. Brain Res. 2014, 1586, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.M.; Kistler, J.P.; Davis, J.M. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hijdra, A.; van Gijn, J.; Nagelkerke, N.J.; Vermeulen, M.; van Crevel, H. Prediction of delayed cerebral ischemia, rebleeding, and outcome after aneurysmal subarachnoid hemorrhage. Stroke J. Cereb. Circ. 1988, 19, 1250–1256. [Google Scholar] [CrossRef]
- Pradilla, G.; Chaichana, K.L.; Hoang, S.; Huang, J.; Tamargo, R.J. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg. Clin. N. Am. 2010, 21, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Altruda, F.; Tolosano, E.; Beringhelli, T.; Fasano, M. Hemoglobin and heme scavenging. IUBMB Life 2005, 57, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Okamoto, S.; Yanamoto, H.; Kikuchi, H. Red blood cells are essential for late vasospasm following experimentally induced subarachnoid hemorrhage in dogs. Neurol. Medico Chir. 1990, 30, 10–15. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Marton, L.S.; Andrus, P.K.; Hall, E.D.; Johns, L.; Sajdak, M. Time course of production of hydroxyl free radical after subarachnoid hemorrhage in dogs. Life Sci. 2004, 75, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Hailer, N.P.; Bechmann, I.; Heizmann, S.; Nitsch, R. Adhesion molecule expression on phagocytic microglial cells following anterograde degeneration of perforant path axons. Hippocampus 1997, 7, 341–349. [Google Scholar] [CrossRef]
- Gallia, G.L.; Tamargo, R.J. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, H.H.; Dacey, R.G., Jr. Molecular keys to the problems of cerebral vasospasm. Neurosurgery 2000, 46, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Pellettieri, L.; Nilsson, B.; Carlsson, C.A.; Nilsson, U. Serum immunocomplexes in patients with subarachnoid hemorrhage. Neurosurgery 1986, 19, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, H.; Shimizu, T. Activated complement components C3a and C4a in cerebrospinal fluid and plasma following subarachnoid hemorrhage. J. Neurosurg. 1989, 71, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, R.; Dinh, Y.R.; Gomis, P. Cerebrovascular inflammation following subarachnoid hemorrhage. Jpn. J. Pharm. 2002, 88, 227–249. [Google Scholar] [CrossRef]
- Hendryk, S.; Jarzab, B.; Josko, J. Increase of the IL-1β and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Neuroendocrinol. Lett. 2004, 25, 141–147. [Google Scholar] [PubMed]
- Mathiesen, T.; Andersson, B.; Loftenius, A.; von Holst, H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J. Neurosurg. 1993, 78, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Okuda, Y.; Kaito, N.; Abe, T. Cytokine production in cerebrospinal fluid after subarachnoid haemorrhage. Neurol. Res. 1995, 17, 106–108. [Google Scholar] [PubMed]
- Vikman, P.; Beg, S.; Khurana, T.S.; Hansen-Schwartz, J.; Edvinsson, L. Gene expression and molecular changes in cerebral arteries following subarachnoid hemorrhage in the rat. J. Neurosurg. 2006, 105, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Sozen, T.; Tsuchiyama, R.; Hasegawa, Y.; Suzuki, H.; Jadhav, V.; Nishizawa, S.; Zhang, J.H. Immunological response in early brain injury after SAH. Acta Neurochir. Suppl. 2011, 110, 57–61. [Google Scholar] [PubMed]
- Chaichana, K.L.; Pradilla, G.; Huang, J.; Tamargo, R.J. Role of inflammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg. 2010, 73, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Sarrafzadeh, A.; Schlenk, F.; Gericke, C.; Vajkoczy, P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2010, 13, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.C.; Davids, A.M.; Brandenburg, S.; Muller, A.; Elke, A.; Magrini, S.; Atangana, E.; Turkowski, K.; Finger, T.; Gutenberg, A.; et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015, 130, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.T.; Schianchi, P.M. Cerebral artery spasm. A histological study at necropsy of the blood vessels in cases of subarachnoid hemorrhage. J. Neurosurg. 1978, 48, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Chyatte, D.; Bruno, G.; Desai, S.; Todor, D.R. Inflammation and intracranial aneurysms. Neurosurgery 1999, 45, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Uekawa, K.; Hasegawa, Y.; Ma, M.; Nakagawa, T.; Katayama, T.; Sueta, D.; Toyama, K.; Kataoka, K.; Koibuchi, N.; Kawano, T.; et al. Rosuvastatin ameliorates early brain injury after subarachnoid hemorrhage via suppression of superoxide formation and nuclear factor-κB activation in rats. J. Stroke Cerebrovasc. Dis. 2014, 23, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Zhang, X.; Wu, Q.; Li, W.; Wang, C.X.; Xie, G.B.; Zhou, X.M.; Shi, J.X.; Zhou, M.L. Astaxanthin offers neuroprotection and reduces neuroinflammation in experimental subarachnoid hemorrhage. J. Surg. Res. 2014, 192, 206–213. [Google Scholar] [CrossRef] [PubMed]
- McGirt, M.J.; Lynch, J.R.; Blessing, R.; Warner, D.S.; Friedman, A.H.; Laskowitz, D.T. Serum von willebrand factor, matrix metalloproteinase-9, and vascular endothelial growth factor levels predict the onset of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery 2002, 51, 1128–1134, discussion 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Feske, S.K.; Simmons, S.L.; Konigsberg, R.G.; Orzell, S.C.; Marckmann, A.; Bourget, G.; Bauer, D.J.; de Jager, P.L.; Du, R.; et al. Elevated peripheral neutrophils and matrix metalloproteinase 9 as biomarkers of functional outco me following subarachnoid hemorrhage. Transl. Stroke Res. 2011, 2, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Feiler, S.; Plesnila, N.; Thal, S.C.; Zausinger, S.; Scholler, K. Contribution of matrix metalloproteinase-9 to cerebral edema and functional outcome following experimental subarachnoid hemorrhage. Cerebrovasc. Dis. 2011, 32, 289–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Song, S.; Sun, G.; Strong, R.; Zhang, J.; Grotta, J.C.; Aronowski, J. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J. Neurosci. Off. J. Soc. Neurosc. 2009, 29, 15819–15827. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Hodapp, B.; Rossol, S.; Bertsch, T.; Schmeck, J.; Schutt, S.; Fritzinger, M.; Horn, P.; Vajkoczy, P.; Kreisel, S.; et al. Inflammatory cytokines in subarachnoid haemorrhage: Association with abnormal blood flow velocities in basal cerebral arteries. J. Neurol. Neurosurg. Psychiatry 2001, 70, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. Off. J. Soc. Neurosc. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Nedergaard, M. Is there a cerebral lymphatic system? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [PubMed]
- Borsody, M.; Burke, A.; Coplin, W.; Miller-Lotan, R.; Levy, A. Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology 2006, 66, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Wong, C.S.; Yeh, C.C.; Borel, C.O. Treatment of cerebral vasospasm after subarachnoid hemorrhage—A review. Acta Anaesthesiol. Taiwan 2004, 42, 215–222. [Google Scholar] [PubMed]
- Rabinstein, A.A.; Weigand, S.; Atkinson, J.L.; Wijdicks, E.F. Patterns of cerebral infarction in aneurysmal subarachnoid hemorrhage. Stroke 2005, 36, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Huang, S.; Ma, L.; Liu, Y.; Li, H.; You, C. Endothelin-receptor antagonists for aneurysmal subarachnoid hemorrhage: An updated meta-analysis of randomized controlled trials. Crit. Care 2012, 16, R198. [Google Scholar] [CrossRef] [PubMed]
- Konczalla, J.; Kashefiolasl, S.; Brawanski, N.; Lescher, S.; Senft, C.; Platz, J.; Seifert, V. Cerebral vasospasm and delayed cerebral infarctions in 225 patients with non-aneurysmal subarachnoid hemorrhage: The underestimated risk of fisher 3 blood distribution. J. Neurointerv. Surg. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bederson, J.B.; Levy, A.L.; Ding, W.H.; Kahn, R.; DiPerna, C.A.; Jenkins, A.L., 3rd; Vallabhajosyula, P. Acute vasoconstriction after subarachnoid hemorrhage. Neurosurgery 1998, 42, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Kassell, N.F.; Sasaki, T.; Colohan, A.R.; Nazar, G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke 1985, 16, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Vora, N. Subarachnoid hemorrhage and inflammation: Bench to bedside and back. Semin. Neurol. 2005, 25, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Altay, T.; Smithason, S.; Volokh, N.; Rasmussen, P.A.; Ransohoff, R.M.; Provencio, J.J. A novel method for subarachnoid hemorrhage to induce vasospasm in mice. J. Neurosci. Methods 2009, 183, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Calisaneller, T.; Ukita, N.; Dumont, A.S.; Kassell, N.F.; Lee, K.S. A murine model of subarachnoid hemorrhage-induced cerebral vasospasm. J. Neurosci. Methods 2003, 123, 89–97. [Google Scholar] [CrossRef]
- Mesis, R.G.; Wang, H.; Lombard, F.W.; Yates, R.; Vitek, M.P.; Borel, C.O.; Warner, D.S.; Laskowitz, D.T. Dissociation between vasospasm and functional improvement in a murine model of subarachnoid hemorrhage. Neurosurg. Focus 2006, 21, E4. [Google Scholar] [CrossRef] [PubMed]
- Parra, A.; McGirt, M.J.; Sheng, H.; Laskowitz, D.T.; Pearlstein, R.D.; Warner, D.S. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: Methodological analysis. Neurol. Res. 2002, 24, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Fujii, K.; Tomura, S.; Ueno, H.; Wada, K.; Otani, N.; Osada, H.; Tomiyama, A. Canine double hemorrhage model of experimental subarachnoid hemorrhage. Acta Neurochir. Suppl. 2015, 120, 347–351. [Google Scholar] [PubMed]
- Springborg, J.B.; Moller, C.; Gideon, P.; Jorgensen, O.S.; Juhler, M.; Olsen, N.V. Erythropoietin in patients with aneurysmal subarachnoid haemorrhage: A double blind randomised clinical trial. Acta Neurochir. 2007, 149, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Fu, X.; Siu, A.; Rasmussen, P.A.; Hazen, S.L.; Ransohoff, R.M. CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit. Care 2010, 12, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Crowley, R.W.; Medel, R.; Kassell, N.F.; Dumont, A.S. New insights into the causes and therapy of cerebral vasospasm following subarachnoid hemorrhage. Drug Discov. Today 2008, 13, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A. The role of microglia and the tlr4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ghosh, S. Toll-like receptor-mediated NF-κB activation: A phylogenetically conserved paradigm in innate immunity. J. Clin. Investig. 2001, 107, 13–19. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, L.A.; Bowie, A.G. The family of five: Tir-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ji, C.; Hu, T.; Wang, Z.; Chen, G. Tamoxifen as an effective neuroprotectant against early brain injury and learning deficits induced by subarachnoid hemorrhage: Possible involvement of inflammatory signaling. J. Neuroinflamm. 2013, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Al-Khindi, T.; Macdonald, R.L.; Schweizer, T.A. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010, 41, e519–e536. [Google Scholar] [CrossRef] [PubMed]
- Kooijman, E.; Nijboer, C.H.; van Velthoven, C.T.; Mol, W.; Dijkhuizen, R.M.; Kesecioglu, J.; Heijnen, C.J. Long-term functional consequences and ongoing cerebral inflammation after subarachnoid hemorrhage in the rat. PLoS ONE 2014, 9, e90584. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Segovia, K.N.; McClure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 2008, 63, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Kotter, M.R. The biology of cns remyelination: The key to therapeutic advances. J. Neurol. 2008, 255 (Suppl. 1), 19–25. [Google Scholar] [CrossRef] [PubMed]
- Buga, A.M.; di Napoli, M.; Popa-Wagner, A. Preclinical models of stroke in aged animals with or without comorbidities: Role of neuroinflammation. Biogerontology 2013, 14, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, M.K.; Crago, E.A.; Conley, Y.P.; Balzer, J.R.; Ren, D.; Ducruet, A.F.; Kochanek, P.M.; Sherwood, P.R.; Poloyac, S.M. 20-hete is associated with unfavorable outcomes in subarachnoid hemorrhage patients. J. Cereb. Blood Flow Metab. 2015, 35, 1515–1512. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, M.K.; Conley, Y.P.; Crago, E.A.; Ren, D.; Sherwood, P.R.; Balzer, J.R.; Poloyac, S.M. Genetic markers in the EET metabolic pathway are associated with outcomes in patients with aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2015, 35, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, S.; Koshy, L.V.; Balan, S.; Easwer, H.V.; Premkumar, S.; Nair, S.; Bhattacharya, R.N.; Alapatt, J.P.; Banerjee, M. Association of versican (VCAN) gene polymorphisms rs251124 and rs2287926 (g428d), with intracranial aneurysm. Meta Gene 2014, 2, 651–660. [Google Scholar] [CrossRef] [PubMed]
- McDowell, M.M.; Ducruet, A.F. The genetics of aneurysms: A complex pathophysiology requiring complex analysis. World Neurosurg. 2015, 83, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Waters, R.J.; Nicoll, J.A. Genetic influences on outcome following acute neurological insults. Curr. Opin. Crit. Care 2005, 11, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.L.; Blackburn, S.; Neal, D.; Mendez, N.V.; Wharton, J.A.; Waters, M.F.; Dore, S. Haptoglobin phenotype predicts the development of focal and global cerebral vasospasm and may influence outcomes after aneurysmal subarachnoid hemorrhage. Proc. Natl. Acad. Sci. USA 2015, 112, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, A.; Unterhuber, V.; Pircher, M.; Schwarz, A.; Gazzeri, R.; Reinert, M.; Widmer, H.R. Psychosocial and neurocognitive performance after spontaneous nonaneurysmal subarachnoid hemorrhage related to the apoe-epsilon4 genotype: A prospective 5-year follow-up study. J. Neurosurg. 2008, 109, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Adamski, M.G.; Golenia, A.; Turaj, W.; Baird, A.E.; Moskala, M.; Dziedzic, T.; Szczudlik, A.; Slowik, A.; Pera, J. The AGTR1 gene A1166C polymorphism as a risk factor and outcome predictor of primary intracerebral and aneurysmal subarachnoid hemorrhages. Neurol. Neurochir. Pol. 2014, 48, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wu, W.; Hu, Y.C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.D.; Ma, B.; Zhu, J.H.; et al. Early release of high-mobility group box 1 (hmgb1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.M.; Chalouhi, N.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Wada, K.; Shimada, K.; Hasan, D.M.; Greig, N.H.; et al. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J. Neuroinflamm. 2014, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ma, Q.; Krafft, P.R.; Hu, Q.; Rolland, W., 2nd; Sherchan, P.; Zhang, J.; Tang, J.; Zhang, J.H. P2x7r/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol. Dis. 2013, 58, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Tosun, C.; Kurland, D.B.; Mehta, R.; Castellani, R.J.; deJong, J.L.; Kwon, M.S.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Inhibition of the SUR1-TRPM4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke 2013, 44, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Sacco, S.; Ornello, R.; Ripa, P.; Pistoia, F.; Carolei, A. Migraine and hemorrhagic stroke: A meta-analysis. Stroke 2013, 44, 3032–3038. [Google Scholar] [CrossRef] [PubMed]
- Dumont, T.; Rughani, A.; Silver, J.; Tranmer, B.I. Diabetes mellitus increases risk of vasospasm following aneurysmal subarachnoid hemorrhage independent of glycemic control. Neurocrit. Care 2009, 11, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.S.; Amidei, C.; Tolentino, J.; Reilly, C.; Macdonald, R.L. Subarachnoid clot volume correlates with age, neurological grade, and blood pressure. Neurosurgery 2007, 60, 259–266, discussion 257–266. [Google Scholar] [CrossRef] [PubMed]
- Mashaly, H.A.; Provencio, J.J. Inflammation as a link between brain injury and heart damage: The model of subarachnoid hemorrhage. Cleve. Clin. J. Med. 2008, 75 (Suppl. 2), S26–S30. [Google Scholar] [CrossRef] [PubMed]
- Acikgoz, S.; Edebali, N.; Barut, F.; Can, M.; Tekin, I.O.; Buyukuysal, C.; Acikgoz, B. Ischemia modified albumin increase indicating cardiac damage after experimental subarachnoid hemorrhage. BMC Neurosci. 2014, 15, 33. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, V.; Flores, R.; Muller, A.; Bi, W.; Peerschke, E.I.; Sehba, F.A. Reduction of neutrophil activity decreases early microvascular injury after subarachnoid haemorrhage. J. Neuroinflamm. 2011, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.C.; Schiffler, J.; Hakiy, N.; Horn, P.; Vajkoczy, P. Functional analysis of pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2012, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M. Models and mechanisms for hippocampal dysfunction in obesity and diabetes. Neuroscience 2015, 309, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Valles, A.; Inoue, W.; Rummel, C.; Luheshi, G.N. Obesity, adipokines and neuroinflammation. Neuropharmacology 2015, 96, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.; Boutin, H.; Jones, M.S.; Denes, A.; McColl, B.W.; Selvarajah, J.R.; Hulme, S.; Georgiou, R.F.; Hinz, R.; Gerhard, A.; et al. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav. Immun. 2011, 25, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Koso, M.; Dizdarevic, K.; Sose-Selimotic, J. Everyday memory in microsurgically treated patients after subarachnoid hemorrhage. J. Clin. Med. Res. 2015, 7, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Wada, K.; Shimada, K.; Makino, H.; Liang, E.I.; Murakami, S.; Kudo, M.; Kitazato, K.T.; Nagahiro, S.; Hashimoto, T. Roles of hypertension in the rupture of intracranial aneurysms. Stroke 2014, 45, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Andresen, M.; Bartek, J., Jr.; Cortsen, M.; Eskesen, V.; Wagner, A. Intra-arterial nimodipine for cerebral vasospasm after subarachnoid haemorrhage: Influence on clinical course and predictors of clinical outcome. Neuroradiol. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Hopkins, S.J.; Hulme, S.; Galea, J.P.; Hoadley, M.; Vail, A.; Hutchinson, P.J.; Grainger, S.; Rothwell, N.J.; King, A.T.; et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: A phase ii randomised controlled trial. J. Neuroinflamm. 2014, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, Q.; Feng, D.; Hu, T.; Fang, Q.; Wang, Z. Expression of NR2B in different brain regions and effect of NR2B antagonism on learning deficits after experimental subarachnoid hemorrhage. Neuroscience 2013, 231, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Boyko, M.; Azab, A.N.; Kuts, R.; Gruenbaum, B.F.; Gruenbaum, S.E.; Melamed, I.; Brotfain, E.; Shapira, Y.; Cesnulis, E.; Zlotnik, A. The neuro-behavioral profile in rats after subarachnoid hemorrhage. Brain Res. 2013, 1491, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, G.K.; Johansson, S.E.; Larsen, C.C.; Samraj, A.K.; Edvinsson, L. Early events triggering delayed vasoconstrictor receptor upregulation and cerebral ischemia after subarachnoid hemorrhage. BMC Neurosci. 2013, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, G.K.; Edvinsson, L. Mek1/2 inhibitor u0126 but not endothelin receptor antagonist clazosentan reduces upregulation of cerebrovascular contractile receptors and delayed cerebral ischemia, and improves outcome after subarachnoid hemorrhage in rats. J. Cereb. Blood Flow Metab. 2015, 35, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Maddahi, A.; Povlsen, G.K.; Edvinsson, L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J. Neuroinflamm. 2012, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.S.; Liu, J.L.; Xie, M.J.; Zhan, Y.; Qu, W.S.; Yu, Z.Y.; Tang, Z.P.; Pan, D.J.; Wang, W. Tamoxifen attenuates inflammatory-mediated damage and improves functional outcome after spinal cord injury in rats. J. Neurochem. 2009, 109, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Guan, J.; Wu, G.; Xi, G.; Keep, R.F.; Hua, Y. Tamoxifen treatment for intracerebral hemorrhage. Acta Neurochir. Suppl. 2011, 111, 271–275. [Google Scholar] [PubMed]
- Osuka, K.; Feustel, P.J.; Mongin, A.A.; Tranmer, B.I.; Kimelberg, H.K. Tamoxifen inhibits nitrotyrosine formation after reversible middle cerebral artery occlusion in the rat. J. Neurochem. 2001, 76, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Fratkins, J.D.; LeBlanc, M.H. Treatment with tamoxifen reduces hypoxic-ischemic brain injury in neonatal rats. Eur. J. Pharmacol. 2004, 484, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Testai, F.D.; Valyi-Nagy, T.; N Pavuluri, M.; Zhai, F.; Nanegrungsunk, D.; Paisansathan, C.; Pelligrino, D.A. Vap-1 blockade prevents subarachnoid hemorrhage-associated cerebrovascular dilating dysfunction via repression of a neutrophil recruitment-related mechanism. Brain Res. 2015, 1603, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Pelligrino, D.A.; Paisansathan, C.; Testai, F.D. Protective role of fingolimod (FTY720) in rats subjected to subarachnoid hemorrhage. J. Neuroinflamm. 2015, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Dumont, A.S.; Calisaneller, T.; Kwan, A.L.; Hwong, S.L.; Lee, K.S. Monoclonal antibody against e selectin attenuates subarachnoid hemorrhage-induced cerebral vasospasm. Surg. Neuro. 2005, 64, 201–205, discussion 205–206. [Google Scholar] [CrossRef] [PubMed]
- Pradilla, G.; Wang, P.P.; Legnani, F.G.; Ogata, L.; Dietsch, G.N.; Tamargo, R.J. Prevention of vasospasm by anti-CD11/CD18 monoclonal antibody therapy following subarachnoid hemorrhage in rabbits. J. Neurosurg. 2004, 101, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Altay, T.; Smithason, S.; Moore, S.K.; Ransohoff, R.M. Depletion of Ly6g/C (+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J. Neuroimmunol. 2011, 232, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of ly6g-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Tosun, C.; Ivanova, S.; Kurland, D.B.; Hong, C.; Radecki, L.; Gisriel, C.; Mehta, R.; Schreibman, D.; Gerzanich, V. Heparin reduces neuroinflammation and transsynaptic neuronal apoptosis in a model of subarachnoid hemorrhage. Transl. Stroke Res. 2012, 3, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kurland, D.B.; Tosun, C.; Pampori, A.; Karimy, J.K.; Caffes, N.M.; Gerzanich, V.; Simard, J.M. Glibenclamide for the treatment of acute CNS injury. Pharmaceuticals 2013, 6, 1287–1303. [Google Scholar] [CrossRef] [PubMed]
- Dumont, A.S.; Dumont, R.J.; Chow, M.M.; Lin, C.L.; Calisaneller, T.; Ley, K.F.; Kassell, N.F.; Lee, K.S. Cerebral vasospasm after subarachnoid hemorrhage: Putative role of inflammation. Neurosurgery 2003, 53, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Polin, R.S.; Bavbek, M.; Shaffrey, M.E.; Billups, K.; Bogaev, C.A.; Kassell, N.F.; Lee, K.S. Detection of soluble e-selectin, icam-1, vcam-1, and l-selectin in the cerebrospinal fluid of patients after subarachnoid hemorrhage. J. Neurosurg. 1998, 89, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tang, K.; Huang, R.Q.; Zhuang, Z.; Cheng, H.L.; Yin, H.X.; Shi, J.X. Therapeutic potential of peroxisome proliferator-activated receptor gamma agonist rosiglitazone in cerebral vasospasm after a rat experimental subarachnoid hemorrhage model. J. Neurol. Sci. 2011, 305, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, X.D.; Zhuang, Z.; Xue, Y.J.; Cheng, H.L.; Yin, H.X.; Shi, J.X. Peroxisome proliferator-activated receptor gamma agonist rosiglitazone attenuates oxyhemoglobin-induced toll-like receptor 4 expression in vascular smooth muscle cells. Brain Res. 2010, 1322, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Germano, A.; Caffo, M.; Angileri, F.F.; Arcadi, F.; Newcomb-Fernandez, J.; Caruso, G.; Meli, F.; Pineda, J.A.; Lewis, S.B.; Wang, K.K.; et al. NMDA receptor antagonist felbamate reduces behavioral deficits and blood-brain barrier permeability changes after experimental subarachnoid hemorrhage in the rat. J. Neurotrauma 2007, 24, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Pradilla, G.; Ruzevick, J.J.; Bender, M.; Edwards, L.; Grossman, R.; Zhao, M.; Rudek, M.A.; Riggins, G.; Levy, A.; et al. A glutamate receptor antagonist, S-4-Carboxyphenylglycine (S-4-CPG), inhibits vasospasm after subarachnoid hemorrhage in haptoglobin 2-2 mice. Neurosurgery 2013, 73, 719–728. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Study | Sex/Species/Age | Model | Drug | Target | Outcome Measures |
|---|---|---|---|---|---|
| (a) Common cisternal SAH model | |||||
| Polvsen & Edvinsson 2015 | Male SD Rats 2–3 months | Cisternal blood infusion | U0126 | MEK1/2 | Neurological score; Behavioral deficits; Cerebral blood flow; Endothelin receptor |
| Maddahi et al., 2012 | Male SD Rats 2–3 months | Cisternal blood infusion | U0126 | MEK1/2 | Neurological score; MAPK pathway; Pro-inflammatory activity; Matrix Metalloproteinase |
| Zhang et al., 2014 | Male SD Rats 2–3 months | Cisternal blood infusion | Astaxanthin | General Anti-oxidant Anti-inflammatory | Neurological score; Blood-brain barrier permeability; Edema; Pro-inflammatory activity; Leukocyte activity; Neuronal cell death |
| Pradilla et al., 2004 | New Zealand White Rabbit 1.5–2.5 kg | Cisternal blood infusion | Antibody | CD11/CD18 | Blood vessel diameter (Vasospasm); Leukocyte activity |
| Provencio et al., 2011 | Male C57 Mice 2–3 months | Cisternal blood infusion | Antibody | Lymphocyte antigen 6 complex locus G6D (Myeloid cells) | Blood vessel diameter (Vasospasm); Leukocyte activity; Behavioral deficits; Microglial response |
| Lin et al., 2005 | Male C57 Mice 30–35 g | Cisternal blood infusion | Antibody | E-Selectin | Blood vessel diameter (Vasospasm); Leukocyte activity |
| Wu et al., 2011 | Male SD Rats 300–350 g | Cisternal blood infusion | Rosiglitazone | Peroxisome proliferator-activated receptor gamma agonist | Blood vessel diameter (Vasospasm); Leukocyte activity; Pro-inflammatory activity |
| Guresir et al., 2013 | Male SD Rats 250–350 g | Cisternal blood infusion | human recombinant Erythropoietin | Erythropoietin receptor | Neurological score; Blood vessel diameter (Vasospasm); Neuronal cell death |
| Germano et al., 2007 | Male SD Rats 250 g | Cisternal blood infusion | Felbamate | N-methyl-d-aspartate receptor antagonist | Blood-brain barrier permeability; Behavioral deficits; Body weight |
| Garzon-Muvdi et al., 2013 | C57 Mice 22–30 g | Cisternal blood infusion | S-4-carboxy-phenylglycine | Glutamate receptor antagonist | Blood vessel diameter (Vasospasm); Leukocyte activity; |
| (b) Clinically-relevant SAH models | |||||
| Xu et al., 2015 | Male SD Rats 2–3 months | Endovascular puncture | LJP-1586 | Semicarbazide-sensitive amine oxidase inhibitor | Neurological score; Leukocyte activity; Microvascular damage |
| Xu et al., 2015 | Male SD Rats 2–3 months | Endovascular puncture | Fingolimod | Sphingosine-1-phosphate receptor modulator | Neurological score; Leukocyte activity; Microvascular damage |
| Simard et al., 2012 | Male Wistar Rats 300–350 g | Entorhinal cortex blood infusion | Heparin | Antithrombin III activator | Demyelination; Neurodegeneration; Pro-inflammatory activity |
| Tosun et al., 2013 | Male Wistar Rats 300–350 g | Entorhinal cortex blood infusion | Glibenclamide | Sur1-Trpm4 channel inhibitor | Neurodegeneration; Behavioral deficits |
| Makino et al., 2012 | Male C57 Mice 2–3 months | Induced hypertension + Elastase injection | Tetracycline Derivatives | Inflammatory Cytokines | Neurological score; Aneurysm rupture at 6 days |
| (c) Clinical Trials for SAH | |||||
| Singh et al., 2014 | Human | Clinical Trials | Anakinra | Interleukin-1 receptor anatagonist | Glasgow outcome score; Blood plasma and cerebral spinal fluid levels of Interleukin-6 between 6 and 24 h |
| Ma et al., 2012 | Human | Clinical Trials | Clazosentan | Endothelin receptor antagonist | Glasgow coma score; Post-SAH vasospasm; Late cerebral ischemia |
| Springborg et al., 2007 | Human | Clinical Trials | Erythropoietin | Erythropoietin receptor | Glasgow outcome score; Transcranial Doppler flow velocity; vasospasm; jugular venous oximetry; Brain injury markers; Blood-brain barrier integrity |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. Int. J. Mol. Sci. 2016, 17, 497. https://doi.org/10.3390/ijms17040497
Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, Huber JD, Rosen CL, Simard JM. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. International Journal of Molecular Sciences. 2016; 17(4):497. https://doi.org/10.3390/ijms17040497
Chicago/Turabian StyleLucke-Wold, Brandon P., Aric F. Logsdon, Branavan Manoranjan, Ryan C. Turner, Evan McConnell, George Edward Vates, Jason D. Huber, Charles L. Rosen, and J. Marc Simard. 2016. "Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review" International Journal of Molecular Sciences 17, no. 4: 497. https://doi.org/10.3390/ijms17040497