Article Text

Abstract
A family with a dominant form of partial GTP cyclohydrolase deficiency is described. Clinical severity varied from mild involvement with complete responsiveness to levodopa to severe dystonia precluding any voluntary activity including talking, progressive contractures, and only partial responsiveness to levodopa. Although there are several possible reasons for intrafamilial variability, any patient with dystonia, the cause of which is not clearly identified, should receive a trial of levodopa.
- GTP cyclohydrolase
- dystonia
- levodopa
- phenotype gene expression
Statistics from Altmetric.com
Cerebral palsy is incurable—although its effects may be alleviated. Great care must be taken when assigning such a diagnosis which implies lifelong disabilities. Nowhere is this more true than when making a diagnosis of dystonic cerebral palsy. Wrongly applied, the chance may be missed of effecting a lifelong cure for a patient with Segawa type levodopa responsive dystonia (DRD).
Responsiveness to levodopa, however, may vary considerably between dystonic syndromes. Segawa type dystonia commonly presents in infancy with dystonic equinus posturing in the feet, sometimes mistakenly diagnosed as a spastic diplegia. Responsiveness to levodopa seems complete and enduring. Less well known is that some cases of secondary dystonia also respond to levodopa.1 2 Hitherto it has been assumed that the degree of responsivity is related to the underlying cause. Identification of the GTP cyclohydrolase I (GTPCH 1) gene as the causative gene in most cases of levodopa responsive dystonia has permitted a broadening of the phenotype from Segawa type DRD at one extreme to partially responsive but severe four limbed dystonia at the other.3 Reported here is a family with the dominant form of partial GTPCH I deficiency within which the phenotype, including responsiveness to levodopa, varied widely to embrace both extremes.
Materials and methods
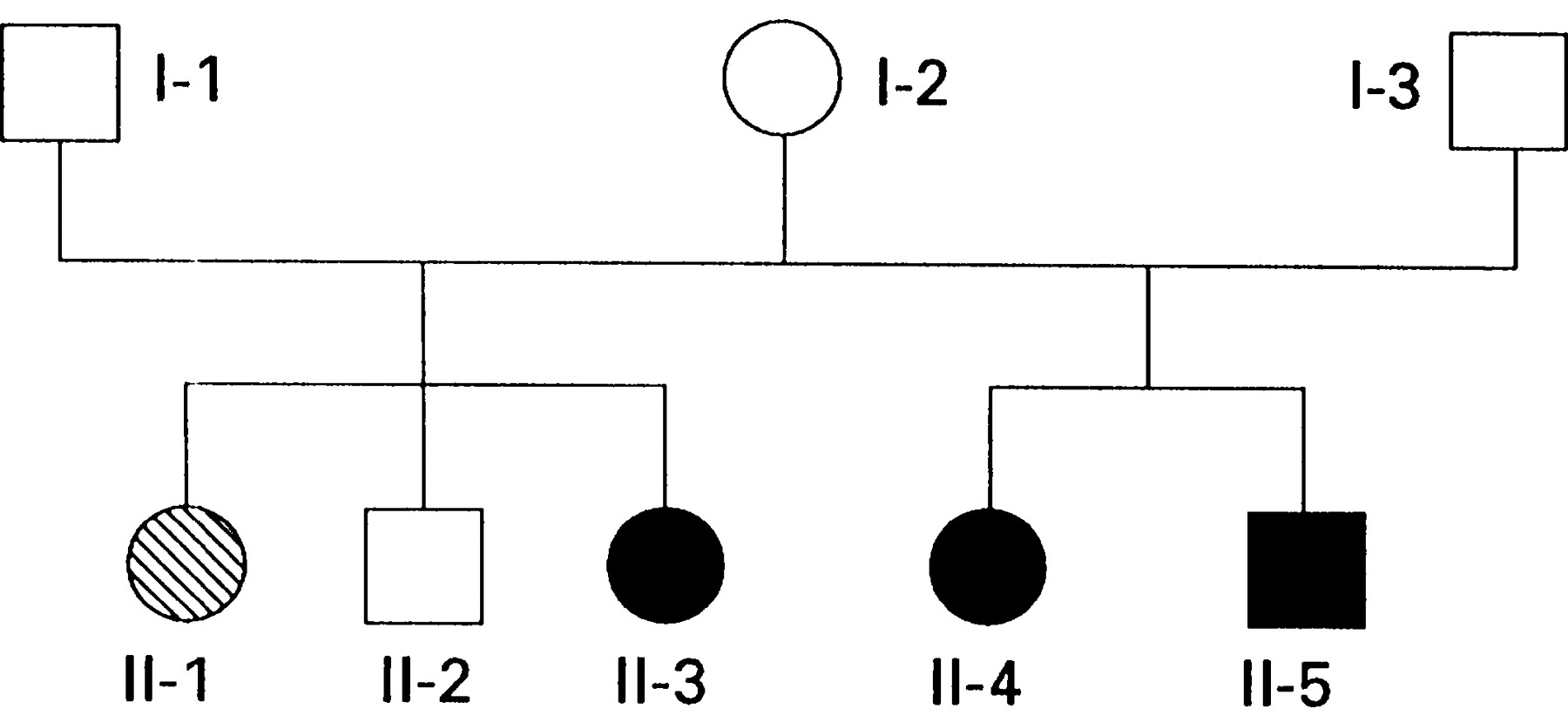
This family first came to our attention through presentation of the 6 year old boy II-5 (fig 1). Subsequently members I-1–3 and II-1−5 have all been examined in detail.

Family tree. The proband is II-5.
CLINICAL DETAILS
The proband II-5 was initially hypotonic, fed well initially, but then experienced increasing feeding problems from about 3 months. His tone then variably increased and, with the age appropriate onset of voluntarily initiated activity, extra pyramidal features mainly of dystonia appeared. A diagnosis of cerebral palsy was made by the local paediatrician. Intelligible words appeared during his second year. His vocabulary developed with effortful articulation, albeit with gradual improvement in speed and clarity. However, this became increasingly difficult for him and he spoke less and less from the age of 5. Seen when aged 6 he was an alert receptive child, apparently intellectually unimpaired with normal social behaviour. He had a pure dystonic syndrome with variable rigidity. His fundi were normal. He was unable to roll purposefully as his arm became trapped beneath him. A form of assisted walking required a lot of pelvic and trunk support. His attempted coordination was best in the morning and least effective after mid-day, until there was again some improvement in the evening. Comprehensive investigations for causes of secondary dystonia were unrevealing. However, CSF neopterin and hydroxyindoleacetic acid (HIAA) were marginally reduced (table), consistent with partial GTPCH 1 deficiency. There was an immediate response to 50 mg levodopa daily with decreased rigidity, resumption of speech, and improved head and trunk control. Increasing the dose to 220 mg daily was accompanied by acquisition of independent sitting, control of individual finger movements, and increase in clarity of speech. However, with disappearance of the dystonia, small amplitude chorea emerged. Further escalation of the dose was prevented by nausea. Six months after starting levodopa, his speech was clear and fluent, he was walking independently and was, for example, able to do up his shoelaces. Further motor progress now seems to rely more on practice than in reduction of motor disorder.
CSF metabolites in II-1, II-4, and II-5
Member II-4 (the older sister of II-5) was likewise hypotonic initially but without feeding difficulties. Although smiling with normal visual behaviour by 6 weeks, her severe motor delay thereafter became more evident. She never acquired independent sitting or standing or useful hand function. Occasional single words first appeared at the age of 18 months, but she did not progress to phrases. The diagnosis of cerebral palsy was made at this time. She developed dysphagia when aged 5. Throughout her childhood and adolescence she was severely handicapped by rigidity. There was, however, clear evidence of diurnal variability with easily intelligible words in the morning which became impossible for her in the afternoon. Her intelligence, as judged by eye pointing and comprehension of language, seemed unimpaired. She developed dislocated hips and a long thorocolumbar kyphoscoliosis. Examined when aged 17, she had a generalised dystonic syndrome with underlying tone, apparently normal between spasms. There were no pyramidal signs. Extraocular movements were preserved. Sensation was normal. Although her CSF neopterin and tetrahydrobiopterin (BH4) concentrations were reduced, her CSF HIAA concentration lay within the normal range (table). Starting initially on 100 mg levodopa a day, she developed troublesome dyskinesias. Restarted at a lower dose, she likewise has begun to improve, being able to tolerate gradually escalating doses of levodopa. She is now able to support her own head, talk more clearly, and feed herself when taking 800 mg levodopa daily. She retains, however, a degree of dystonia with some fixed flexion contractures as well as developing some unwanted movements.
Member II-3, the half sister of II-4 and II-5, developed symptoms of stiffness and toe walking aged 7 years. She underwent a bilateral achilles tenotomy aged 11. Examined when aged 22, she had a stiff legged gait with reduced step length and internal foot progression angle. She was least symptomatic shortly after waking. She has had a complete and enduring remission of all symptoms on 150 mg levodopa a day.
II-1, the sister of II-3 (and half sister of II-4 and II-5) developed Friedreich’s ataxia with the onset of progressive ataxia when aged 13. Her CSF neopterin, biopterin, and HIAA lie within the normal range although her HVA is somewhat lower (table). She lacks the GTPCH I gene mutation of her affected family members and is homozygous for the GAA expansion in intron 1 of the Friedreich’s ataxia gene.
I-1, I-2, and I-3 are all clinically unaffected.
CSF monoamine metabolites and individual pterin species were measured in II-1, II-4, and II-5 using established high performance liquid chromatography (HPLC) methods.4 The GTPCH I gene has also been sequenced in these members. This family is the family Hu, included as one of the five previously reported with point mutations of the GTPCH I gene.4 Members II-4 and II-5 have a point mutation on codon 224 of the gene changing lysine to arginine (AAA –> AAG).
Discussion
GTPCH I is the initial and rate limiting enzyme de novo in BH4 synthesis from GTP. Tetrahydrobiopterin is an essential cofactor for phenylalanine, tyrosine, and tryptophan hydroxylases. Tyrosine hydroxylase in turn is the initial and rate limiting enzyme in dopamine synthesis. A deficiency in GTPCH I activity therefore readily explains both the movement disorder and the changes in CSF HIAA, neopterin, and BH4 concentrations (fig 2).

{kind=link}
{kind=link}
Pathways of biopterin, dopamine, and serotonin synthesis.
The autosomal recessive form of GTPCH I deficiency causes no detectable enzyme activity5 6 Patients are normally detected on newborn screening for phenylketonuria but most already have symptoms of dopamine deficiency (personal observations).6 CSF concentrations of homovanillic acid (HVA), 5HIAA, neopterin, and BH4 are all markedly reduced (personal observations).5Heterozygotes do not manifest clinical symptoms and have 30% to 50% residual enzyme activity.5 By contrast, it has been suggested that in the dominant form of GTPCH I deficiency described here residual enzyme activities are 2% to 20% of normal (instead of the 50% expected) implying that the mutation causes dysfunction of the normal gene. In recessively inherited GTPCH I deficiency, heterozygotes have 30% to 46% activity and are normal.7 Biochemical abnormalities in CSF are also more subtle in the dominant form. The only consistent abnormality in the affected patients here is a modest reduction in CSF neopterin to 22% to 30% of mean reference values, similar to that previously found.8 9 A reduction of CSF BH4 was found in II-4 but not II-5 and a reduction in HIAA was found in II-5 but not II-4. Surprisingly, CSF HVA was normal in II-4 and II-5 at a time when both had symptoms of dopamine deficiency, but was reduced in II-1. These findings may be explained by the activity of dihydropteridine reductase maintaining BH4 concentrations, the replenishment of dopamine stores during motor inactivity and because lumbar CSF acts as a sump. The reduction of CSF HVA in II-1, who has Friedreich’s ataxia, is consistent with the low concentrations found in other inherited ataxias.10
The dominant form described here is one of the causes of levodopa responsive dystonia—the other being recessively inherited tyrosine hydroxylase deficiency.11 12 The DRD gene has been localised13 to chromosome 14q in the region between 14q11–24.3. The GTPCH I gene has subsequently been mapped to the same region and mutations of this gene have been shown to cause DRD.3 7 Patient II-3 seems to have a pure form of DRD. Although her DNA has not been available for sequencing, it is likely that she shares the same GTPCH I mutation as her more severely affected half brother and sister (members II-4 and II-5). This being so, the importance of the family lies not only in confirming that GTPCH I deficiency can cause pure Segawa type DRD, but also within the same family, partially levodopa responsive dystonia. This intrafamilial variability may be due to the effect of other modifier genes or varying concentrations of other substances such as nerve growth factor known to enhance expression of GTPCH I. Other possibilities include independent mutations of other genes in the synthetic pathway of BH4 or dopamine or undescribed environmental factors affecting dopa production. Although on direct questioning, a modest degree of diurnal variability was prominent in the most severely affected patients, this may not always be the case in levodopa responsive syndromes. It has been clear for some time that a trial of levodopa is warranted in dystonic syndromes and may prevent the need for an extensive and costly search for causes of secondary dystonia. The gratifying response to levodopa in II-5 as compared with the less complete response by II-4 may be related to the more prolonged period of the condition in the second together with development of subsequent contractures. Direct GTPCH I gene sequencing will allow prenatal testing, testing of asymptomatic individuals if requested, as well of course as direct diagnosis when a gene abnormality has been identified in an affected relative. Although a complete remission of symptoms is to be hoped for, this may not always be the case but a partial response in our experience gives considerable relief. Whichever is the case, we suggest that all children with an extrapyramidal motor disorder should be given a trial of levodopa unless there is incontrovertible evidence of hypoxic ischaemic encephalopathy.