Article Text

Abstract
Background The pathogenesis of multifocal motor neuropathy (MMN) has yet to be established. MMN patients often carry anti-GM1 IgM antibodies, suggesting an autoimmune process involving complement. Intravenous immunoglobulin (IVIG) is the first line treatment, but its action mechanism is unknown.
Objective To test whether anti-GM1 IgM antibodies in MMN sera activate complement, inducing and propagating the disease and whether IVIG inhibits complement activation, resulting in clinical improvement.
Methods Sera with anti-GM1 IgM but not IgG or IgA reactivity were obtained from 13 patients with MMN. We tested whether their anti-GM1 IgM antibodies produced complement component deposits on GM1-coated microtiter plates and whether IVIG blocks such deposition.
Results C1q, C4b, C3b and C5b-9 were deposited on GM1-coated wells. Their depositions were highly correlated with anti-GM1 IgM antibody titre. IVIG reduced the deposition of these complement components dose-dependently.
Conclusions Anti-GM1 IgM antibodies bound to GM1 and activated complement in vitro. The results together with earlier data from our group suggest that IgM-induced, complement-mediated injury occurs at the nodes of Ranvier in peripheral motor nerves and generates conduction block and muscle weakness. In vitro IVIG inhibited this type of complement activation, suggesting that in vivo, the resulting reduction in membrane attack complex-mediated damage leads to improved muscle strength.
- Anti-GM1 antibody
- complement
- conduction block
- intravenous immunoglobulin
- multifocal motor neuropathy
- ganglioside
- guillain-barre syndrome
- immunology
- neuroimmunology
- neuropathy
Statistics from Altmetric.com
- Anti-GM1 antibody
- complement
- conduction block
- intravenous immunoglobulin
- multifocal motor neuropathy
- ganglioside
- guillain-barre syndrome
- immunology
- neuroimmunology
- neuropathy
Multifocal motor neuropathy (MMN) is characterised by progressive, predominantly distal asymmetric limb weakness that mostly affects the upper limbs with minimal or no sensory impairment.1 Nerve conduction studies have found multifocal persistent partial conduction blocks on motor but not on sensory nerves. Serum IgM antibodies to ganglioside GM1 have been detected in 30% to 80% of MMN patients, but their pathogenic role remains unknown. Cumulative evidence, however, supports the hypothesis that autoantibodies bind to gangliosides, activate the classical complement system pathway and induce nerve injury by incorporation of the complement membrane attack complex (C5b-9) in peripheral motor nerves.2 3 MMN patients do not usually respond to steroids or plasma exchange and their conditions may worsen if given these treatments; therefore, intravenous immunoglobulin (IVIG) is the first-line treatment.4 Because the immunopathogenesis of MMN is unknown, the IVIG action mechanism has yet to be clarified. Our primary aim to clarify MMN pathogenesis in part was to test whether the anti-GM1 IgM antibodies efficiently activate via the classical complement pathway. Based on the findings, a second aim was to investigate whether IVIG blocks complement deposition mediated by those autoantibodies in order to develop more effective MMN treatments.
Materials and methods
Blood samples
Serum samples from individual patients were obtained from university and district general hospitals throughout Japan for anti-ganglioside antibody assays as described elsewhere.5 A written informed consent for the use in other experimental studies was obtained from every patient. Samples with anti-GM1 IgM but no detectable IgG or IgA reactivity were selected from 13 patients and from another ten who had no anti-GM1 antibodies but fulfilled the published diagnostic criteria for MMN.4 Each serum sample had been obtained before immunotherapy, and most care was given to handling of samples in order to avoid damaging proteins. Samples were thawed only once before use. Anti-asialo GM1 and anti-GD1b IgM antibodies, respectively, were positive for all and five of the 13 patients; whereas, anti-GD1a and anti-GQ1b antibodies were negative for all of them. Normal human sera (NHS) samples obtained in bulk from five healthy subjects were frozen in 1-ml aliquots. Aliquots of the patients' sera and NHS were stored at –70°C until used. The hospital ethics committee approved the performance of this study.
Human plasma protein concentrates for in vitro studies
Kenketu Glovenin®-I (Glovenin®; Nihon Pharmaceutical, Tokyo, Japan), Kenketu Venilon®-I (Venilon®; KAKETSUKEN, Kumamoto, Japan) and Kenketsu Jochu-globulin “KAKETSUKEN” ® (F(ab′)2; KAKETSUKEN) were dissolved in sterile water according to the manufacturer's instructions (50 mg/ml). Glovenin®was preferred in the majority of experiments because it is the accepted IVIG therapy for MMN and chronic inflammatory demyelinating polyneuropathy (CIDP) under Japanese insurance. S-sulphonated IVIG, Venilon®, is registered for treatment of Guillain–Barré syndrome (GBS) in Japan; whereas, pepsin-treated IVIG, F(ab′)2 is not registered for treatment of neurological conditions. Human serum albumin (HSA; Kenketsu Albumin 20 “KAKETSUKEN”, KAKETSUKEN) was the control protein used. IVIGs and HSA samples were dialysed against phosphate-buffered saline (PBS, pH 7.4) before use.
IgM binding and complement deposition assays
Between each step, the microtiter plates (Sumilon MS-7296F, Sumitomo, Tokyo, Japan) were washed six times with PBS containing 0.05% Tween 20. Individual wells were coated in quadruplicate with 10 ng of GM1, GD1a or GQ1b in PBS at 37°C for 1 h. Wells were blocked with PBS containing 4% casein. Each ganglioside was purified from its bovine brain ganglioside mixture by Q-Sepharose (Pharmacia, Uppsala, Sweden) column chromatography.6 In the first incubation, sera diluted 1:1000 or more with PBS containing 4% casein were added to individual wells, with or without ganglioside, then incubated at 4°C overnight. For the IgM binding step, sera were deliberately not heat-inactivated because of concern regarding aggregation of immunoglobulins which would have falsified experimental results. Instead, a high dilution that rendered complement deposition negligible was used. As a source of complement, NHS diluted 1:50 or more with PBS containing 4% casein was added at the second step, and the plates incubated at 37°C for 1 h. To detect the C4b and C3b depositions on each well, peroxidase-conjugated anti-human C4 (CEDARLANE Laboratories, Ontario, Canada) and anti-human C3 (1:8000; MP Biomedicals LLC, Solon, Ohio, USA) antibodies, respectively, were diluted 1:100 and 1:8000 with PBS containing 0.5% casein then added to the plates and incubated at 37°C for 1 h. To detect C1q deposition, biotin-conjugated anti-human C1q antibodies (1:1000; abcam, Tokyo, Japan) were added. The plates were incubated after which peroxidase-conjugated streptavidin (1:2000; Sigma, St. Louis, Missouri, USA) was added. To detect C5b-9 deposition, rabbit anti-human C5b-9 antibodies (1:4000; abcam) were added, the plates incubated, then peroxidase-conjugated anti-rabbit IgG antibodies (1:1000; Amersham Biosciences, Little Chalfont, UK) were added. Each plate next was developed with enzyme substrate for 15 min at room temperature. Optical densities of the ganglioside-free wells were subtracted from densities of ganglioside-coated wells. None of the peroxidase conjugated-antibodies and streptavidin reacted with GM1.
To test whether IVIG blocks complement deposition mediated by bound anti-GM1 antibodies, 50 μl of diluted NHS was mixed with an equal volume of IVIG in each well, and then the wells were incubated at 37°C for 1 h. Treatment of patients with IVIG (2 g/kg of body weight) is reported to induce serum IgG levels of 32–50 mg/ml in vivo,7 but most of the in vitro assays were done with 10 mg/ml of IVIG. Each assay then followed standard procedure using those antibodies against human C1q, C4, C3 or C5b-9.
To investigate whether IVIG inhibits the binding of anti-GM1 antibodies to GM1, 50 μl of diluted sera was mixed in each well with an equal volume of IVIG, and the plates were incubated at 4°C overnight. To determine whether IVIG displaces anti-GM1 antibodies once-bound to GM1, sera were incubated at 4°C overnight, the plates washed and exposed to IVIG at 37°C for 1 h. Each assay then followed standard procedure using those antibodies against human IgM or C3.
Statistics
Spearman's rank test was used for the correlations, two-way repeated measures ANOVA for treatment and concentration and Wilcoxon's signed rank test for comparison of values before and after treatment (Prism®, GraphPad Software, La Jolla, California, USA). Differences were considered statistically significant if the p value was less than 0.05.
Results
Activated complement deposition mediated by GM1-reacted anti-GM1 IgM antibodies
Deposition of C1q, the first complement reacting with IgM-containing immune complexes, and of C4, another component of the classical complement pathway, as well as deposition of C3, was highly correlated to anti-GM1 IgM antibody titres (C1q, r=0.79, p=0.0013; C4b, r=0.85, p=0.0003; C3b, r=0.93, p<0.0001; figure 1A), indicative that anti-GM1 antibodies from MMN patients can mediate complement deposition. No C3b deposition was detected after incubation of MMN sera with no GM1 reactivity (n=10), amyotrophic lateral sclerosis sera (n=10), chronic inflammatory demyelinating polyneuropathy sera (n=10) or healthy control sera (n=10). No C3b was deposited on GD1a- or GQ1b-coated wells when MMN sera with GM1 reactivity were used. C5b-9 is the final product of complement activation, and there was a strong correlation between C3b and C5b-9 deposition (r=0.82, p=0.034; figure 1B) although only seven sera were available for the latter evaluation. These in vitro results indicate that complement deposition through C1 to C9 can be mediated by GM1-reacted anti-GM1 IgM antibodies.

Activated complement deposition on microtiter plates mediated by serum anti-GM1 antibodies in MMN. (A) Correlation of anti-GM1 IgM antibody titres with C1q (n=13, r=0.79, p=0.0013), C4b (n=13, r=0.85, p=0.0003) and C3b deposition (n=13, r=0.93, p<0.0001). (B) Correlation between C3b and C5b-9 deposition (n=7, r=0.82, p=0.034). In panels A and B, patients' sera were diluted 1:1000. Normal human sera (1:50 dilution) were the complement source. Results are presented as optical densities (OD) at 492 nm. Individual values are the mean (SEM) of four wells.
Inhibition of activated complement deposition by IVIG
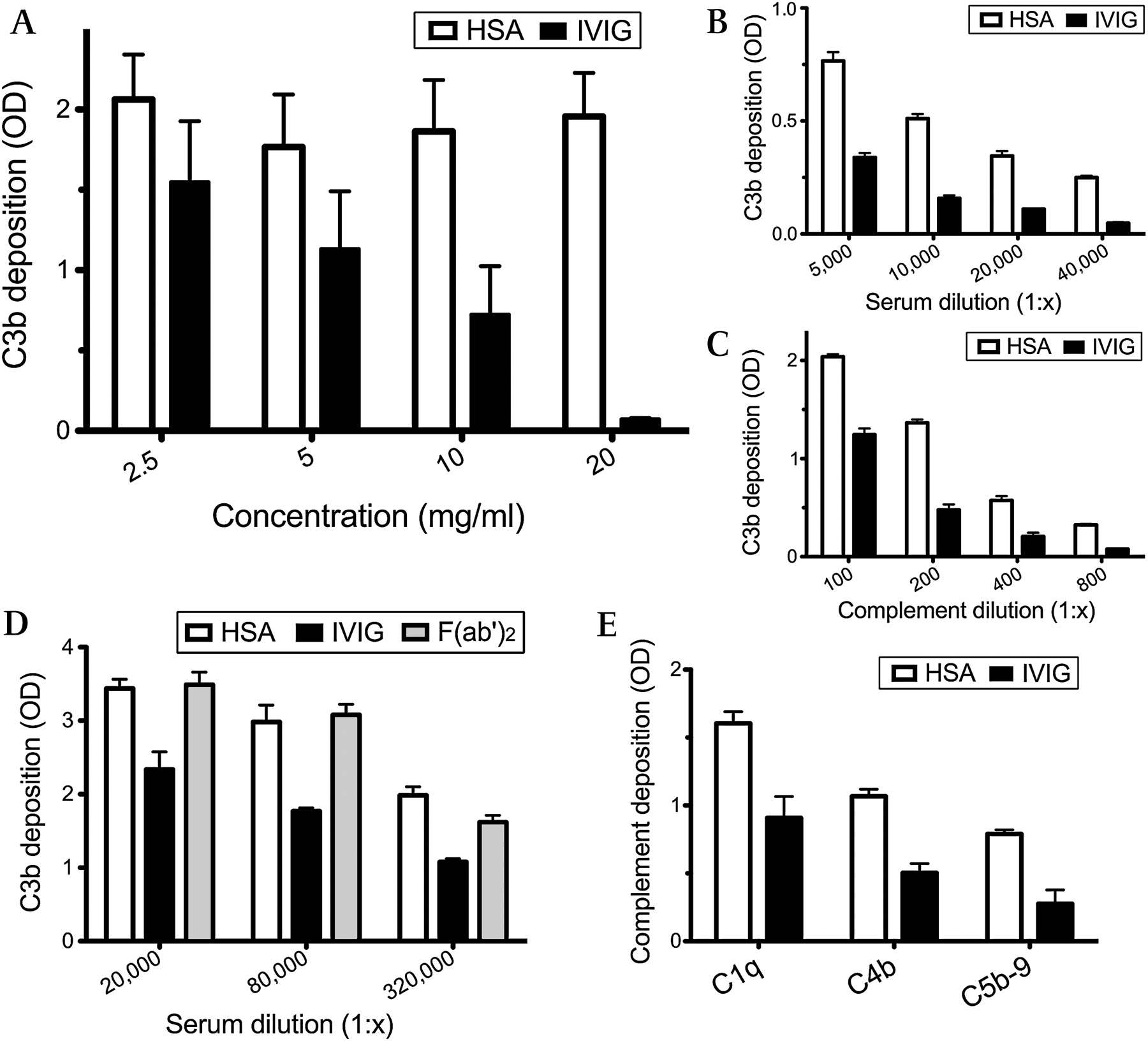
In in vitro inhibition assays, IVIG-mediated blocking of complement deposition varied based on three parameters: (1) IVIG dose, (2) autoantibody dose and (3) complement dose. Figure 2A shows that IVIG dose-dependently reduced C3b deposition in the eight MMN sera with a 1:5000 or higher titre of anti-GM1 IgM antibodies, whereas HSA had no effect on complement deposition (p=0.017). The C3b deposition inhibitory effect did not differ significantly for the three lots of Glovenin® and two lots of Venilon® (data not shown). C3b deposition was reduced at higher dilutions of patient serum and complement source. IVIG reduction exceeded the effect of complement dilution. Figure 2B,C shows representative results for serum with high anti-GM1 IgM antibody titre. In contrast, an immunoglobulin preparation containing pepsin-digested IgG, F(ab′)2, had no inhibitory effect (figure 2D), suggesting that the Fc portion of the IgG molecules is necessary for inhibitory activity. In addition to C3b deposition, IVIG also blocked the depositions of C1q, C4b and C5b-9 (figure 2E). Similar results were obtained for sera from three different patients (figure 2D and E; representative results).

Complement deposition on microtiter plates modulated by IVIG. (A) Dose-dependent inhibition of C3b deposition by IVIG compared with human serum albumin (HSA; p=0.017). Samples from eight patients who had high anti-GM1 IgM antibody titres were diluted 1:1000. Results are given as optical densities (OD) at 492 nm. Individual values are the mean±SEM of the eight samples. (B, C) Reduction of C3b deposition in the presence of constant amounts of IVIG (10 mg/ml) and serial dilutions of a patient's serum (B) or complement source (C). (D) Inhibition of C3b deposition by F(ab′)2 (10 mg/ml) and intact IgG. F(ab′)2 had inhibitory capacities similar to HSA, indicative that the Fc portion of IgG has a role in the inhibition of complement activation. (E) Decreases in C1q, C4b and C5b-9 depositions by IVIG (10 mg/ml). In panels C to E, serum from a patient who had high anti-GM1 antibody titre (1:128 000 or more) was diluted 1:10 000. In panels A, B, D and E, normal human sera (1:100 dilution) were the complement source. In panels B to E, individual values are the mean (SEM) of three independent experiments.
Anti-idiotype blocking and displacement activity by IVIG
IVIG's blocking effect on antibody binding to GM1 was studied in three MMN sera samples. In those assays, IVIG was added to the MMN sera at the first incubation step. Results of IgM binding to GM1 (data not shown) as well as of C3b deposition (figure 3A) showed that IVIG was a more effective blocker than HSA but was less effective than F(ab′)2. These results suggest that anti-idiotypic antibodies block the binding of anti-GM1 antibodies to GM1. Figure 3A shows representative serum results for a patient.

{kind=link}
{kind=link}
{kind=link}
Complement deposition modulated by the anti-idiotype and displacement activity of IVIG. (A) Blockage of anti-GM1 antibody binding to GM1 by IVIG and F(ab′)2 (10 mg/ml) determined by C3b deposition. (B) Displacement of GM1-bound anti-GM1 IgM by IVIG. Patient serum was first incubated with coated GM1. In a second incubation, the complexes formed were exposed to increased doses of IVIG. Remaining complexes subsequently were detected by assessing deposited C3 with normal human sera as the complement source. Antibodies once bound to GM1 apparently are not displaced by IVIG. (C) No displacement of anti-GM1 antibodies once bound to GM1 by IVIG (10 mg/ml) despite the presence of anti-idiotype activity and complement inhibition on the same plate. In panels A to C, serum obtained from a patient who had high anti-GM1 antibody titre (1:128 000 or more) was diluted 1:10 000. Normal human sera (1:100 dilution) was the complement source. Results are shown as OD at 492 nm. Individual values are the mean (SEM) of three independent experiments.
To investigate whether IVIG can displace anti-GM1 antibodies bound to GM1, IVIG was added after MMN samples were incubated with GM1. C3b deposition (figure 3B) and IgM binding shows that IVIG did not displace the bound anti-GM1 antibodies. Furthermore, at a diluted condition on the same plate, IVIG showed anti-idiotype activity and complement inhibition, but no displacement (figure 3C). Complement inhibition was more prominent than anti-idiotype activity. Similar results were obtained for two other patients' sera. Figure 3B,C shows representative results for one serum sample.
Discussion
Possible pathogenic role of anti-GM1 IgM antibodies in the development of conduction block
The pathophysiology of MMN has yet to be clarified.1 Anti-GM1 IgM antibody is a useful MMN diagnostic marker, but its pathogenic role has yet to be established. GM1/anti-GM1 IgG and IgM-induced formation of C5b-9 in vitro previously was studied in sera from patients with presumably immune-mediated neuropathies and sera from various controls.8 A significant correlation between the anti-GM1 serum level (IgM plus IgG) and respective complement-activating capacities was found. In our study, parameters were restricted to the IgM isotype of anti-GM1 antibodies in MMN sera and resulting complement activation. The latter was studied at various steps of the complement cascade. Results suggest that complement activation occurs from C1 to C9 when solid-phase GM1 reacts with IgM antibodies in MMN patients' sera. Indeed, C1q, C4b, C3b (the breakdown products of C4 and of C3) and C5b-9 were deposited on GM1-anti-GM1 complexes, and the extent of deposition was highly correlated with serum anti-GM1 IgM antibody content. Interestingly, certain anti-GM1 antibodies exert potent complement activation-mediated neuropathogenic effects in mice, including morphological damage at living terminal motor axons, leading to a block of synaptic transmission.9
The pathogenic role of autoantibodies against GM1 is better known in acute motor axonal neuropathy (AMAN), an axonal subtype of GBS. GM1 is a target molecule for IgG antibodies in AMAN patients.10 IgG, C3b and C5b-9 are deposited at the nodes of Ranvier in the spinal anterior roots. Some GBS patients who carry anti-GM1 antibodies show multifocal conduction block.11 Sensitisation of rabbits with GM1 induces production of anti-GM1 IgM and IgG antibodies. These autoantibodies bind at the nodes of Ranvier in the spinal anterior roots and activate complement as deduced from C3b and C5b-9 being deposited at the nodes then at the paranodes.2 Autoantibody binding and complement activation result in the disappearance of voltage-gated sodium channel clusters and detachment of paranodal myelin terminal loops. These changes may produce conduction block and muscle weakness in AMAN.
A similar pathophysiological mechanism may function in the development of MMN because GM1 as well as GD1a, GM1b and GalNAc-GD1a1 10 12 are common target molecules for autoantibodies in AMAN and MMN. Unlike in AMAN, pathophysiological abnormalities at the conduction block site in MMN may arise from depolarisation or hyperpolarization.13 14 The pathomechanism remains in question because in one study injection of serum from a patient with MMN associated with anti-GM1 IgM antibodies together with fresh human complement to rat sciatic nerve resulted in conduction block and immunoglobulin deposits at the nodes of Ranvier,15 but this could not be confirmed.16 Pathological alteration of the axon predominated over myelin pathology17; whereas the presence of depositions of IgM or complement has yet to be shown in MMN patients. In addition, a main difference between MMN and GBS is the lack of response to plasma exchange,1 a fact discussed below. Further studies are required to show whether anti-GM1 IgM antibodies activate complement and induce conduction block and muscle weakness in MMN.
Possible action mechanism of IVIG in MMN associated with anti-GM1 antibodies
The IVIG action mechanism has not been clarified in MMN because of the condition's unknown pathogenesis. As discussed above, MMN associated with anti-GM1 antibodies may be caused by complement activation subsequent to autoantibody binding to GM1. Two studies have shown that anti-ganglioside antibodies mediate complement-dependent neural injury and that IVIG prevents complement activation.18 19 One concluded that in an ex vivo mouse neuromuscular junction model IVIG inhibited the binding of anti-GQ1b antibodies to GQ1b in sera from patients with Fisher syndrome, thereby preventing complement activation and subsequent pathological effects.18 In the in vitro assay of the other study, the conclusion was that in anti-GM1 or anti-GD1a sera from GBS patients, the IVIG-mediated protection mechanism included the action of anti-idiotypic antibodies and the downregulation of complement activation.19 No similar studies, however, have been published for MMN.
Our in vitro study showed that IVIG could prevent complement activation mediated by anti-GM1 IgM antibodies from MMN patients. A previous publication reported that IVIG and its F(ab')2 fragment dose-dependently inhibited the binding of anti-GM1 IgM antibodies to GM1 in MMN sera, evidence of an anti-idiotypic effect in vitro.20 Our study findings confirm this earlier observation. Additionally, we showed that an anti-idiotypic effect resulted in inhibition of C3b deposition. Unlike in another previous study that showed IVIG displaced anti-GQ1b antibodies,18 in our in vitro assay, IVIG did not displace anti-GM1 antibodies once they had bound to GM1.
The following beneficial action mechanisms of IVIG have been described: (1) IVIG-mediated scavenging of activated complement,21 (2) antibody-mediated binding of anaphylatoxins22 and (3) inhibition of C3 activation via the amplification loop23 24 in pathologies in which complement activation is involved. In our study, we assessed inhibition of complement deposition to surface-bound complexes of GM1/anti-GM1 IgM. The test system did not allow distinguishing between action mechanisms (1) and (3). IVIG, however, dose-dependently reduced the depositions of all the complement components assessed.
The inflammatory effect of IVIG mediated via control of activation of complement rises a challenging question: Why is plasmaphaeresis effective in GBS but not in MMN? We like to point to the following facts: Plasmaphaeresis removes complement and pathological antibodies, however, does not add strong regulatory components thereafter. Recovery of local complement is quite rapid as there is a local complement synthesis. Besides the abovementioned anti-inflammatory effect (1) immunoglobulins through the V-connected network have in addition an immediate and direct influence on biding of pathological autoantibodies, as shown here and elsewhere; (2) interacting with T- and B-cell receptors, they can influence long-term the immunoglobulin repertoire and (3) they can influence dendritic cell maturation.25 In GBS, an acute disease not being in a steady-state, the mild regulatory effects of plasmaphaeresis apparently are sufficient to mediate an improvement. In MMN, however, a chronic condition having established some level of steady state, the more powerful immunomodulatory effect of IVIG is needed to achieve an improvement.
A better understanding of the IVIG action mechanism will aid in developing more effective treatments for MMN. Moreover, more rational treatment may make use of complement inhibitors. The murine model of Fisher syndrome has been treated successfully with eculizumab, a humanised monoclonal antibody that binds to and blocks cleavage of C5.26 Several serine proteases activate the classical and alternative complement system pathways, and a synthetic serine protease inhibitor, nafamostat mesilate, inhibits complement deposition and prevents sodium channel cluster disruption in the AMAN rabbit model.27 In our in vitro study, nafamostat mesilate blocked the C3b deposition mediated by anti-GM1 IgM antibodies in MMN (data not shown). Complement inhibitors, therefore, may provide optional treatment of MMN.
In conclusion, our findings proved that there is a binding of pathogenic anti-GM1 autoantibodies from MMN sera to the target antigen GM1 in vitro. This suggests a similar effect happening during MMN progression. As a consequence of formation of antigen-antibody complexes, local complement activation occurs from C1q to C5b-9, the latter representing the tissue-damaging membrane attack complex. The results presented support the view of multi-site concomitant IVIG action benefitting MMN patients. They indicate that IVIG counteracts the immune complex-initiated, complement-mediated tissue damage activation at various stages; by anti-idiotypic activity, as well as inhibition of complement deposition by scavenging nascent C3b and, probably more important, by specific inhibition of the alternative pathway of C3 convertase assembly.23 24 As a consequence of reduced C3 activation, generation of the membrane attack complex and tissue damage are reduced. In other words, IVIG may inhibit complement activation and in situ deposition, thereby leading to improvement of muscle strength.
References
Footnotes
Competing interests None.
Ethics approval This study was conducted with the approval of the Dokkyo Medical University, Tochigi, Japan.
Provenance and peer review Not commissioned; externally peer reviewed.