Article Text

Abstract
Blood-brain barrier (BBB) disruption has long been recognised as an important early feature of multiple sclerosis (MS) pathology. Traditionally, this has been seen as a by-product of the myelin-specific immune response. Here, we consider whether vascular changes instead play a central role in disease pathogenesis, rather than representing a secondary effect of neuroinflammation or neurodegeneration. Importantly, this is not necessarily mutually exclusive from current hypotheses. Vascular pathology in a genetically predisposed individual, influenced by environmental factors such as pathogens, hypovitaminosis D and smoking, may be a critical initiator of a series of events including hypoxia, protein deposition and immune cell egress that allows the development of a CNS-specific immune response and the classical pathological and clinical hallmarks of disease. We review the changes that occur in BBB function and cerebral perfusion in patients with MS and highlight genetic and environmental risk factors that, in addition to modulating immune function, may also converge to act on the vasculature. Further context is provided by contrasting these changes with other neurological diseases in which there is also BBB malfunction, and highlighting current disease-modifying therapies that may also have an effect on the BBB. Indeed, in reframing current evidence in this model, the vasculature could become an important therapeutic target in MS.
- Multiple Sclerosis
- Blood-brain Barrier
- Neuropathology
- Immunology
- Cerebrovascular
Statistics from Altmetric.com
Introduction
Since Charcot’s description towards the end of the 19th century, multiple sclerosis (MS) has been characterised by demyelinating plaques of the central nervous system (CNS). While these inflammatory lesions have now been well studied, there is still much debate surrounding the aetiology and early pathogenesis of the disease. Classical transplantation experiments by Medawar first highlighted the concept of immune privilege in the CNS, and the blood-brain barrier (BBB) has long been thought to play a key role in this by regulating leucocyte movement into the CNS. When formulating a hypothesis for the pathogenesis of MS 30 years ago, Poser highlighted that one of the four factors needed for the production of central demyelinating lesions was ‘a subsequent alteration of the BBB resulting from diverse mechanisms including trauma or a second, immune-mediated event’. While more complex immune surveillance of the CNS is now recognised, and there are newer ‘outside-in’ and ‘inside-out’ theories of MS pathogenesis,1 most work has remained focused on an autoimmune model whereby the immune system is somehow activated in the periphery leading to immune infiltration of the CNS and inflammatory demyelination.
Neuroimaging studies using gadolinium (Gd) enhancement and postmortem findings of perivascular immune infiltrates have established that BBB disruption is an early feature of MS pathogenesis.2 Indeed, more recent MRI studies suggest that global BBB disruption is an early feature that predicts conversion of optic neuritis to MS,3 and longitudinal studies in mice have shown that BBB disruption occurs before immune cell infiltration in focal lesions.4 However until now, disruption of the BBB has been seen as a step that is required for MS lesion formation but not at the core of its aetiology.
Other excellent reviews have cautiously hinted at the fact that BBB dysfunction might precede immune cell infiltration, but otherwise focus on how the BBB is disrupted by the immune system, rather than directly asking if there is some other mechanism for BBB disruption.2 Here, we propose a new model wherein genetic and environmental associations, which have been predominantly studied in terms of immune function, might also converge to act on the BBB, highlighting an important role for vessel pathology in the disease. We suggest that this BBB disruption may be an important early step that leads to the initiation of a CNS-specific immune response, which ultimately causes the pathological hallmarks of MS. We herein present a detailed review of this topic and include key references in the main text. However, given the breadth of the topic, additional important references can be found in the online supplementary material.
The BBB in health
The concept of a specialised boundary between the circulation and brain parenchyma originated from the observation that dyes injected into the veins of animals stained all tissues but the CNS. The BBB is now recognised as a tightly regulated cellular barrier which maintains CNS tissue homeostasis by facilitating controlled metabolic exchanges while preventing the entry of pathogens, peripheral immune cells and other potentially neurotoxic factors. Maintaining this complex functionality is contingent on multifaceted and dynamic interactions between endothelial cells, pericytes, astrocytes and the basement membrane which together form the neurovascular unit (NVU).
The endothelial cell (EC) forms the innermost layer of the NVU. Compared with their counterparts in the peripheral circulation, ECs in the BBB have reduced pinocytic activity and a lack of transendothelial fenestrations,5 but a larger repertoire of specific carrier and transport systems to facilitate more controlled, although more energy-expensive, transcellular flux. The paracellular route is almost entirely sealed by tight junctions (TJ) and adherens junctions (AJ). TJs comprise complexes of transmembrane proteins including occludin, claudins and junctional adhesion molecules (JAMs). The cytoplasmic tails of these transmembrane proteins mediate signalling pathways essential for TJ regulation and EC survival,5 while the extracellular loops bind those on neighbouring cells to create a ‘zip-lock’ barrier, which is crucial in restricting the paracellular movement of most bloodborne substances. AJs appear to fill the remainder of the paracellular cleft but are unlikely to contribute significantly to BBB permeability as expression of components is normal in postmortem MS brains.
Pericytes are localised within the endothelial basement membrane (BM) and are uniquely placed to influence all components of the NVU through N-cadherin-dependent binding and secretion of diffusible factors and extracellular matrix components. Pericytes are more abundant in the CNS than any other organ system and are critical in maintaining cerebral capillary function in both health and disease. Analysis of viable pericyte-deficient mice has shown that pericytes are necessary for the formation of the BBB in embryogenesis, that absolute pericyte coverage influences BBB permeability and that pericytes are able to suppress the rate of transcytosis and the expression of leucocyte adhesion molecules in ECs.6 They are also the effectors in neurovascular coupling, the phenomenon by which capillary diameter, and therefore perfusion, is regulated by neuronal activity7 via signals from astrocytes. Interestingly, pericytes may also have antigen-presenting features due to expression of MHC II and phagocytic properties and, more recently, multipotent stem cell activity which permits differentiation into both neural and vascular cell lineages.
Astrocytic end-feet form the outermost layer of the BBB, the glia limitans, which covers 99% of the surface of brain microvessels. Formation of the glia limitans coincides with the development of barrier properties in the BBB and astrocyte-EC interactions are thought to be crucial in regulating the BBB phenotype. This includes control of angiogenesis, transporter protein expression and TJ protein expression and morphology.2 This is exemplified in experiments where barrier properties, including TJ integrity, can be reinstated in monocultured ECs by coculture with astrocytes or astrocyte-conditioned media.8 The glia limitans also receives contributions from neurons and microglia, although the functional relevance of this is yet to be fully characterised.
There are two BMs found in the BBB: the endothelial BM supports ECs and pericytes, while the closely associated parenchymal BM supports the glia limitans. Between these two BMs lies pia-lined perivascular spaces, also known as Virchow Robin spaces (VRSs), which contain interstitial fluid and perivascular macrophages. This system is thought to be involved in ‘lymphatic’ drainage of the brain to cervical lymph nodes, which may play a role in the initiation of an immune response against myelin in MS.
The BBB in MS
A substantial body of radiological and pathological evidence is increasingly showing BBB disruption as an early feature of MS.
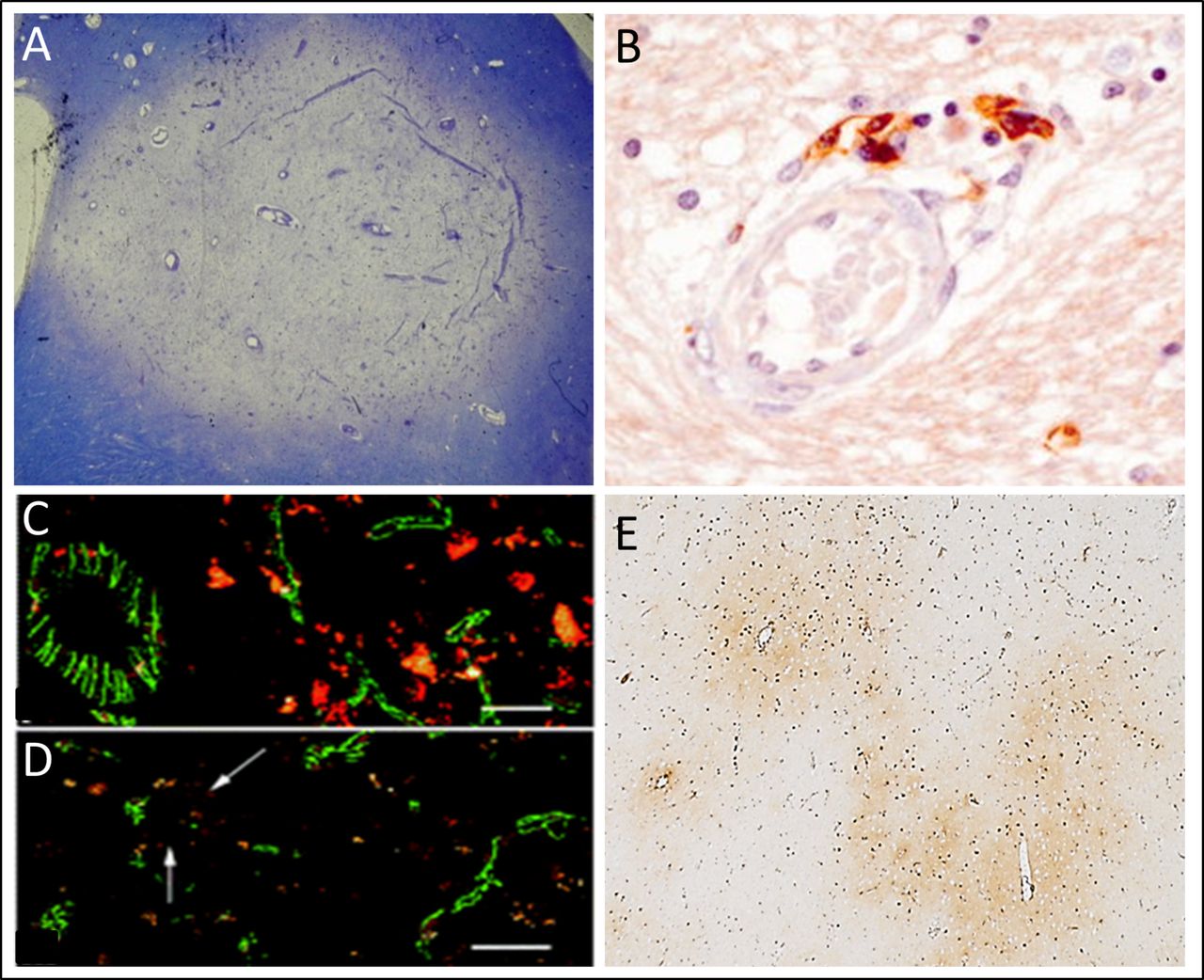
Regarding radiographic evidence, one of the clearest indications of BBB disruption is the presence of Gd-enhancing lesions on MRI. Gd-enhancement is a putative marker of BBB disruption, is associated with active inflammation in lesions and is consequently one of the key diagnostic criteria for an acute MS lesion. An increasing amount of evidence suggests that this disruption of the BBB is not restricted to Gd-enhancing lesions. MRI changes appear in the normal-appearing white matter before enhancing lesions,9 subtle BBB disruption is seen in non-enhancing areas10 and BBB disruption precedes symptoms and other MRI signs. These BBB changes appear to extend to the very earliest stages of disease as global BBB disruption is present at the onset of optic neuritis in patients who go on to develop MS.3 In terms of histopathological evidence, seminal characterisation of the MS lesion by Dawson in 1916 recognised that myelin breakdown invariably originated around parenchymal blood vessels11 (figure 1A, B). It is intuitive that disruption of barrier function is a prerequisite for the entry of an inflammatory infiltrate into perivascular areas and it follows that this barrier disruption is likely to be one of the early events in lesion formation. This is supported by evidence of fibrinogen deposition, a marker of endothelial permeability, in developing lesions.10

Multiple sclerosis (MS) lesions are associated with vascular abnormalities. Acute MS lesions (A) are typically centred around vessels where perivascular inflammation (B) is an early feature. Tight junctional abnormalities (C, D) and the egress of high molecular serum proteins (e.g. fibrin(ogen)) from the vasculature into the brain parenchyma (E) occur in early, relapsing-remitting and late progressive MS. These pathological features put vascular abnormalities at the centre-stage of lesion pathogenesis. (A) A Luxol Fast Blue Cresyl Violet stained section of an acute periventricular white matter lesion. (B) A perivascular MS lesion immunostained for CD8, with permission from Prineas and Parratt.11 (C and D) Immunofluorescence and confocal microscopy sections from normal cortical grey matter of patients with primary progressive MS illustrating regions of (C) normal ZO-1 reactivity (green) and (D) abnormal Zonula occludens-1 (ZO-1) reactivity (arrows indicate abnormal tight junctions). With permission from Leech et al.12 (E) Fibrin(ogen) in the motor cortex of a patient with progressive MS.
These findings are complemented by evidence of maladaptive changes in the components of the NVU. ECs display chemokines on their luminal surface and upregulate key adhesion molecules to facilitate transcellular immune cell migration.2 The paracellular route is also compromised due to changes in the expression and organisation of TJs (figure 1C, D). TJ abnormalities are seen across the brain in both relapsing-remitting and progressive stages of MS12 being most common in active lesions. Furthermore, these TJ abnormalities are associated with fibrinogen leak and perivascular astrogliosis.2 Astrocytes themselves appear to exhibit a number of degenerative changes including a failure to upregulate AQP4 and retraction of astrocytic end-feet from the glia limitans. The underlying BM appears irregular and discontinuous in active lesions and deposition of BM components is found within active lesions.13 Interestingly, knockout of matrix metalloproteinase (MMP)-2 and MMP-9, T cell-derived MMPs which cleave BM components, in mice prevents infiltration beyond the parenchymal BM and conveys resistance to Experimental Autoimmune Encephalomyelitis (EAE).14 There is also a putative role for the intervening VRS in the inflammatory response in MS—neuroimaging studies show that the numbers and volumes of VRSs are increased in patients with MS compared with healthy controls.15 Pathological changes in pericytes in MS are yet to be characterised and may yield interesting findings given their importance in maintaining vascular integrity and regulating capillary perfusion.
There may be topographical differences in BBB phenotype in MS which could in turn contribute to the substantial topographical variability encountered in the disease. There has been increasing attention on the idea that the subpial demyelination seen in the cortex may be more related to diffusible factors associated with meningeal inflammation than a cellular infiltrate secondary to BBB disruption. This idea has developed from observations that Gd-enhancement is rarely seen in the cortex, that cortical demyelination and axonal loss are not associated with a cellular infiltrate or overt signs of BBB disruption,16 and that tertiary-lymphoid like follicles in the subarachnoid space associate with diffuse meningeal inflammation, cortical demyelination and accelerated clinical decline in secondary progressive MS (SPMS).17 However, this does not provide the full picture. First, enhancement with standard doses of Gd has a relatively low sensitivity in detecting BBB disruption.10 Second, pathological studies are often limited by the late-stage disease manifest in the postmortem brains—studies on samples from early stages show that cortical demyelination is, in fact, associated with a cellular infiltrate.18 Furthermore, recent evidence from our group shows widespread fibrin(ogen) deposition throughout the cortex in progressive disease (Figure 1E, Figure 2A). Most importantly, it is important to recognise that the subarachnoid and the VRS are in fact contiguous and therefore, one cannot focus on meningeal inflammation without considering the impact of BBB disturbance and perivascular inflammation. At the other end of the spectrum, it is interesting to note that the blood-spinal cord barrier is thought to be more permeable than the BBB given that spinal cord symptoms are common early in the disease course and become a predominant feature in the progressive phase.
There has also been considerable debate over the BBB phenotype in progressive disease. It has been suggested that the BBB regains integrity in the progressive phase with ‘compartmentalised inflammation’ occurring behind a sealed BBB.19 This idea has been used to justify the absence of Gd-enhancing lesions on MRI and the failure of current therapies in the progressive stages of disease. There is, however, little evidence to support this idea. Furthermore, fibrin(ogen) deposition across the MS cortex correlates with neuronal loss20 which, taken with the TJ abnormalities seen in normal-appearing grey matter in progressive MS,12 could implicate BBB disruption in progressive disease (figure 2B).
Cerebral perfusion in MS
With regard to our hypothesis, it is important to appreciate the recognised role of the vasculature on a more macroscopic level. MRI studies show global cerebral hypoperfusion in all stages of MS,21 which is significant for two reasons. First, this hypoperfusion may be a primary phenomenon, and therefore key to MS pathogenesis, as it presents before any grey matter volume loss in early relapsing-remitting MS (RRMS)22 and occurs out of proportion to any reduced metabolic demand associated with axonal loss. Second, this hypoperfusion may lead to hypoxia, which is becoming increasingly recognised as a mechanism for tissue injury in MS. Appreciation for this arose from the observation that a subset of early lesions show hypoxic injury similar to white matter stroke with predominant oligodendrocyte degeneration, selective loss of myelin-associated glycoprotein and upregulation of nuclear HIF-1α23 24 (figure 2C, D). Oligodendrocytes are especially susceptible to hypoxia in models of hypoxic-ischaemic injury25 pointing to a potentially causal role for hypoxia in the early MS lesion. There is also a growing appreciation that oxidative stress secondary to chronic hypoxia may be an important mechanism for neurodegeneration and axonal loss in MS. In fact, tissue hypoxia is directly related to neurological deficit in the EAE mouse model and is mitigated by oxygen administration26 (Figure 2E). Cells of the NVU are also affected. Pericytes undergo contraction and death in rigour in response to neuronal ischaemia leading to irreversible capillary constriction and further BBB damage.7 In vitro experiments show that neuronal ischaemia also triggers Vascular Endothelial Growth Factor(VEGF-A) secretion from astrocytes which disrupts TJs and increases endothelial permeability. Most importantly, this hypoperfusion may ultimately lead to focal plaque formation as recent work has shown that MS lesions are most common in watershed areas of low arterial blood supply.27 28

Consequences of vascular abnormalities in multiple sclerosis (MS). Total fibrin(ogen) deposition, a surrogate marker of blood-brain barrier disruption, is markedly increased in the progressive MS cortex compared with non-neurological controls (A), and is associated with significant reduction in neuronal density in the functionally relevant layer 5 (B). Vascular changes may also contribute to a hypoxic milieu (C and D) which, in the animal model EAE, associates with more severe disability scores (E). (A) Total fibrin(ogen) deposition in different layers of the motor cortex in MS cases (black bars) vs controls (white bars); (B) Influence of fibrin(ogen) on layer 5 neuronal density in MS. A and B from reference 20.(C and D) HIF-1α expression, which reflects hypoxia, shown to be focused in an area adjacent to an active demyelinating lesion in concentric sclerosis. At higher magnification (D), cytoplasmic expression and nuclear translocation shown in cells resembling oligodendrocytes. With permission from reference 24.(E) Oxygen concentrations measured using an oxygen probe in lumbar spinal cord grey matter in rats with varying EAE severity. With permission from reference 26.
It is, however, still unclear as to what the initiating factor for the hypoxia might be. It is important to note that hypoxia can either be ‘true’, that is, secondary to a deficiency in oxygen delivery, or ‘virtual’, due to mitochondrial dysfunction and an inability to use delivered oxygen. It is likely that a combination of these is active in MS and is associated with the global hypoperfusion evident in the disease. Nitric oxide and superoxide produced in innate immune activation induces virtual neuronal hypoxia and would lead to a secondary reduction in perfusion through the neurovascular coupling phenomenon. This is supported by a recent study showing that early MS lesions occur at watershed sites when the innate immune system is activated by injection of LPS into the spinal cord. Early hypoxia and increased production of nitric oxide and superoxide precede oligodendrocyte loss and demyelination, and this can be significantly reduced by normobaric oxygen administration.27 True hypoxia is associated with hypoperfusion and reduced oxygen delivery which could either be primary or secondary to BBB dysfunction. With regard to a primary disturbance in flow, there is a recognised association between small vessel disease and MS. Vascular comorbidity is associated with more rapid disability progression29 and there is an increased risk of ischaemic stroke and myocardial infarction in the first years following a diagnosis of MS.30 While there are no prospective studies to elucidate the direction of causality, it could be possible that hypoperfusion secondary to macrovascular disease is a pathogenic factor in MS. With regard to a primary BBB dysfunction, astrocytes and pericytes are the key regulators of capillary blood flow and maladaptive changes in these cells may therefore cause the reduction in flow. It has been suggested that endothelin-1 (ET-1) released by reactive astrocytes may mediate the hypoperfusion seen in MS as the 20% deficit in cerebral blood flow seen in patients with MS can be normalised with an infusion of an ET-1 antagonist.31 It is still very unclear whether the causal step for hypoxia is focal changes in flow itself, primary BBB dysfunction or increased metabolic demands of inflamed CNS tissue and unpicking these relationships may be crucial in understanding the pathogenesis of the early lesion. Regardless of the cause, these findings suggest that cerebral hypoperfusion and tissue hypoxia may be important factors in MS pathogenesis and are synergistic with BBB disturbance in a vasculopathic model.
This first section illustrates the importance of BBB disruption and vascular changes in MS pathogenesis. It is now important to consider the potential causal factors of these disturbances in MS.
Genetic influences
A genetic risk for developing MS has long been recognised, and has been well reviewed elsewhere.32 33 Family studies have shown variable levels of heritability, with a recent large Swedish cohort indicating a sevenfold increased risk among siblings. Over 100 discrete loci have been implicated in genome-wide association studies (GWAS).32 Most of these have been implicated and studied in terms of immune function. A pathological immune response is necessary for MS pathology, but these genes may also have independent roles in early BBB dysfunction. No study to date has considered this. Interestingly, there is overlap between genetic associations for MS and cardiovascular disease,34 which may implicate a genetic influence on the vasculature or endothelium.
A literature review examining associations between the 20 highest ranked MS-associated genes on DisGeNET35 and the BBB demonstrates a potential for their impact on components of the BBB (table 1; online supplementary table 1). For example, ApoE has been implicated in BBB integrity in Alzheimer’s disease (AD). But while two SNPs in ApoE had previously been implicated in MS, a more recent GWAS study provided good evidence that these are in fact not implicated in MS susceptibility. One of the strongest genetic associations with MS is the HLA-DRB1*15:01 allele which encodes a class II MHC molecule, whose primary role in MS is in presentation of CNS antigens by antigen-presenting cells.32 There is no evidence of a direct influence of this allele on the BBB aside from an association with ALCAM polymorphisms. Furthermore, recent work in our group did not show increased cortical fibrin(ogen) deposition in brains of patients with HLA-DRB1*15:01 positive MS compared with controls.20 This finding suggests that there may not be an association between HLA-DRB1*15:01 and BBB dysfunction, although it may be confounded by abnormalities in fibrin(ogen) clearance seen in MS.
Potential associations of MS-associated genes with BBB function. The 20 highest ranking genes on DisGeNET were searched for in PubMed with the keywords ‘blood brain barrier’, ‘endothelial’, ‘endothelium’, ‘astrocyte’, ‘pericyte’ and ‘tight junction’. Results were reviewed and the most relevant possible associations are discussed. *Data from UniProt (http://www.uniprot.org/). All references for this table are included in the online supplementary table 1
There are potential links between genes implicated in MS and specific components of the NVU (table 1; online supplementary table 1). Their roles in endothelial function and leucocyte migration have been most widely studied. For example, IL7R, IL-2RA and CD58 are expressed by human endothelial cells, cytokines such as IL1b and LIGHT have been shown to activate the endothelium, and there are multiple genetic associations with adhesion molecules implicated in leucocyte transmigration, for example, ICAM-1, CD6 and CBLB. However, it is important to note that some of these studies do not specifically show a role in endothelial cells in the CNS. Astrocytes are also linked functionally to implicated genes: IL7R is expressed in mice astrocytes, CIITA is thought to play a role in astrocytes’ ability to function as antigen-presenting cells, and one study suggests that IL-1b abolishes astrocytes’ protective effect on BBB integrity. The only potential genetic link with pericytes identified in our search was a study in a mouse tumour model showing that intratumoral LIGHT improves vascular integrity through increased pericyte contractility. This exemplifies the issues with identifying potential genetic associations between the BBB and MS—many MS-associated genes are immune-related pleiotropic genes, often studied in animal models of NVU components, and of different phenotypes outside the CNS. Furthermore, whether some of these associations truly exist has been debated between studies of different sample sizes and populations. No specific polymorphism or genetic variation has been linked to NVU function in MS as of yet, but it is clearly an area that warrants further investigation.
To date, no firm genetic influence on the BBB has been shown in MS. However, in the model we are proposing, effects on the BBB and immune system could be synergistic factors in causing disease. A dysfunctional BBB in an individual with a genetically predisposed immune system could result in an increased likelihood of developing MS and a greater disease burden.
Environmental influences
Environmental factors undoubtedly play a role in MS pathogenesis.33 Here we argue that these classically associated factors—including Vitamin D exposure, smoking and pathogen exposure—converge on the BBB to cause its disruption, as well as impacting on inflammatory demyelination and axonal degeneration through immunomodulatory effects.
Vitamin D
A role for hypovitaminosis D in MS pathology is now well established.33 36 Epidemiological studies show a latitude gradient and a month-of-birth effect correlating MS risk with levels of sun exposure. Patients with MS typically have a lower serum 25(OH)D level and vitamin D supplementation decreases the risk of developing MS.36 Higher vitamin D levels, however, are associated with decreased axonal injury in MS.
Understandably, theories surrounding the role of vitamin D in pathogenesis of MS have focused on immunomodulatory effects, the roles of vitamin D response elements in HLA-DRB1*15 and non-MHC-related genes and vitamin D receptors on lymphocytes.36 However, to our knowledge, no research has examined the effects of vitamin D on the BBB in MS. Vitamin D deficiency has been linked with increased cardiovascular risk due to endothelial dysfunction.37 Furthermore, vitamin D has been shown to play a direct role in controlling BBB permeability in a mouse model of ischaemic injury through a VDR-NF-κB-MMP-9-mediated pathway.38 In another rat model of SAH, intranasal vitamin D3 has been shown to be protective at the BBB through upregulation of the glycoprotein osteopontin.39 Osteopontin is found in MS plaques and induces relapses in EAE models of RRMS by inducing pro-inflammatory reactions and inhibiting apoptosis of T cells.40 Clearly this conflicts, and most likely represent different osteopontin responses in traumatic and inflammatory disease. But while osteopontin appears to have mostly pro-inflammatory effects, it also has cytoprotective and anti-inflammatory roles, and its role in the BBB in MS could be more complex than is currently understood. Osteopontin is expressed by the endothelium in MS,40 maybe this reflects BBB protection in spite of its pro-inflammatory effects. While these studies are in animal models of diseases clearly distinct from MS, they do illustrate a potential link between vitamin D and BBB function which may be relevant to MS.
Smoking
Inhaling tobacco smoke is strongly associated with a dose-dependent increase in the risk of developing MS, a worse prognosis of disease and an increased probability of developing resistance to biological treatments.33 Astonishingly, one Swedish population study attributed 20.4% of MS cases to smoke exposure41while another showed that each year of smoking after initial diagnosis accelerated conversion of RRMS to secondary progressive MS by 4.7%.42 Patients with MS should be strongly advised to quit.
Again, the role of smoking in modulating immune-mediated pathology in MS dominates current research.33 The effects of smoking in more global vascular disease are well established, but research into a direct influence of tobacco smoke on the BBB remains sparse.43 There is an increased rate of small vessel brain disease in smokers, and a link has been demonstrated between small vessel disease and MS.44 The nicotine, free radicals and nitric oxide associated with smoking have all been shown to have a disruptive effect on tight junctions and endothelial cell function in the brain.43 In an in vitro BBB model, tobacco smoke directly reduced BBB viability through a pro-inflammatory response mediated by IL-6 and TNF-α and MMP-2 and MMP-9 metalloproteinase activation of endothelial cells, astrocytes and monocytes. Notably, when there was concurrent cessation of blood flow, the effect on BBB disruption was doubled.45 Again this study was in a model distinct from MS, but levels of metalloproteinases including MMP-2 and MMP-9 are increased in perivascular cuffs in acute MS lesions, serum MMP-9 levels have been correlated with levels of Gd-enhancement46 and MMP cleavage at the parenchymal BM is associated with EAE pathology.14 In addition, there may be a synergistic effect with cerebral hypoperfusion. Smoking could conceivably cause both true hypoxia, through vasoconstriction and carbon monoxide, and virtual hypoxia, through nitric oxide generation, which can lead to a cascade of events which ultimately results in injury to CNS tissue and the BBB. Therefore, a feasible, but as of yet unproven, link exists between smoking, BBB disruption and MS pathogenesis.
Pathogens and microorganisms
There have been debates around the role of an infectious cause of MS since the epidemics in the Faroe Islands among World War II troops. In the 1980s, a link between chronic sinusitis and MS relapses was established by Derek Gay, in the absence of any isolated pathogen, which is especially pertinent in light of newer ‘nose-to-brain’ hypotheses of MS aetiology.47 Further longitudinal studies have shown a correlation between clinical systemic infection and MS relapses. Included in the long list of infectious agents implicated in MS is Epstein-Barr virus (EBV), cytomegalovirus, human herpes virus 6, Chlamydia pneumoniae and Clostridium perfringens.33 In most no direct causal link has been established, and while epidemiological evidence might show an association, the topic remains controversial. Symptomatic EBV infection arguably has the most backing33 as individuals who have had infectious mononucleosis have an increased risk of MS.
Most pathogen associations are thought to be solely immunomodulatory, in concordance with variations on the hygiene hypothesis implicated in other autoimmune diseases. But more recent evidence suggests additional roles of pathogens in direct disruption of the BBB. Perhaps most interestingly, EBV has been shown to be able to directly infect brain microvessel ECs in vitro and cause an inflammatory response.48 The authors further suggest a model wherein only a small proportion of ECs would need to be infected to result in disease, which may account for lack of virus detection in plaques and variable detection in diseased brains. Another more controversial pathogen implicated in MS is C. perfringens. It has been isolated in one patient at first presentation of MS, and its epsilon toxin, a key cause of enterotoxaemia in ruminants, has been shown to bind to the BBB and cause disruption. The authors of one study suggest that the toxin might have independent effects on neurodegeneration and BBB disruption49 in a similar vein to other MS risk factors where there might be independent effects on disruption of the BBB and formation of pathological lesions.
There are further putative roles of bacterial products in influencing neuroinflammation and the BBB. For example, peptidoglycan has been detected in antigen-presenting cells in MS lesions.50 Injection of LPS into dorsal column white matter in rats results in demyelinating lesions at nearby but distinct watershed zones, with this effect decreased if the rats’ inspired oxygen was increased.27 The NVU is a key part of the innate immune system, and in particular cerebral endothelial cells express TLR2 which senses peptidoglycan, stimulation of which increases BBB permeability. Innate immunity could trigger localised hypoxia and BBB disruption that could lead to CNS-specific immune responses in MS. However, these bacterial components are ubiquitous among the microbiome so it is arguably difficult to envisage an MS-specific mechanism.
A role for microorganisms in the development of acute MS lesions and relapses has dominated current research, but an effect of chronic infection in progression and neurodegeneration is also possible. The Power group have demonstrated that there is an altered CNS microbiome in chronic MS pathology, and show that levels of peptidoglycan correlate with demyelination and inflammation in MS.51 Since peptidoglycan may influence the BBB through the innate immune system, as discussed above, there is a potential link between chronic dysbiosis and BBB disruption that could contribute to progressive disease. The gut microbiome might also influence chronic MS pathology. One 16S rRNA sequencing study examining the gut microbiomes of patients with MS with a mean disease length of 12.8 years showed decreased levels of butyrate-producing Butyricimonas species compared with controls.52 Butyrate produced by bacteria or given orally has been shown to be protective of the BBB in both MS and non-MS models,52 which suggests that a lower level of butyrate-producing gut commensals in progressive MS could contribute to chronic BBB dysfunction.
BBB disruption in other neurological diseases
BBB dysfunction is a prominent feature in a range of other neurological diseases and, while the BBB changes in these diseases are often thought to be secondary to a primary pathology, a subset of these carry BBB disruption as a potentially key pathogenic step. Examination of the BBB dysfunction seen in such diseases is valuable in contextualising the changes we see in MS.
Neuromyelitis optica (NMO) is an autoimmune astrocytopathy, which shares some clinical and pathological features with MS. IgG to astrocytic AQP4 causes activation of complement and extravasation of granulocytes leading to BBB compromise and focal demyelination. NMO lesions are characteristically found in a circumventricular distribution—areas which are more readily accessed via the CSF than the BBB. A recent paper from the Lucchinetti group suggests that anti-AQP4 IgG may in fact gain initial access to the CNS through the choroid plexus and the CSF rather than the more tightly regulated BBB.53 Once in the CSF, IgG can bind AQP4 expressed on the basolateral surface of ependymal cells and the pial glia limitans and thereby access the brain parenchyma. While it has not been the focus of this review, this blood-CSF-brain pathway represents another important barrier between the systemic circulation and the brain parenchyma and may also contribute to the pathogenesis of MS, especially given that the subarachnoid and VRSs are contiguous.
Neuropsychiatric symptoms are manifest in 40% of those with systemic lupus erythematosus (SLE) and illustrate the necessity of BBB breakdown in allowing immune factors access to the CNS. SLE is associated with a plethora of plasma neurotoxic antibodies, including those against the NMDA receptor, but it is unclear how these enter the CNS. Neuropsychiatric symptoms arise independently of systemic levels of antibody and are more likely related to compromise of the BBB54 through factors such as infection, including EBV and CMV, and smoking. These are risk factors for both MS and SLE, although not specifically for neuropsychiatric symptoms, and point to the potential importance of environmental factors in altering the BBB phenotype and disease process. Furthermore, irregularities in the walls of small vessels due to vasculitis and intramural platelet deposition55 may lead to altered flow, hypoxic tissue damage and further BBB disruption.
BBB disruption is also seen in neurodegenerative diseases. A neurovascular hypothesis of AD argues that the BBB is the converging point of pathogenic events leading to dementia.56 The theory posits that BBB dysfunction leads to both reduced perfusion and beta-amyloid accumulation in the brain parenchyma, through increased deposition and reduced clearance across the damaged BBB. These factors together are thought to lead to neurodegeneration. In support of this idea, beta-amyloid deposition in microvessel walls and BBB disruption in the hippocampus are present from early stages of the disease, and maladaptive changes are seen in all cells of the NVU. It is unclear why a degenerative rather than an inflammatory phenotype results from this dysfunction. There are a number of likely contributing factors. First, immune senescence associated with advancing age may dampen any neuroinflammation that may result from a damaged BBB and a consequently exposed CNS. This phenomenon is evident in MS itself in which levels of neuroinflammation in older cases return to the level of age-matched controls.19 Second, peripheral immune dysfunction, as seen in MS, NMO and SLE, may be required for BBB disturbance to result in neuroinflammation. Third, it is possible that the quality of the BBB disruption is different between AD and MS. For example, cerebral microbleeds are present in AD but not in MS and the incidence and severity of AD lesions in MS is no higher than age-matched controls57 suggesting that the BBB disruption seen in MS and AD is distinct.
Actions of disease-modifying drugs on the NVU
A wide range of disease-modifying drugs are available for RRMS. These lead to a reduction in radiographic lesions and clinical relapses, but there remains no treatment for progressive disease and certainly no cure. Immunomodulatory effects acting on downstream immune-mediated pathology is the main mechanism of action of all currently used therapies. Additional effects on the BBB may have been overlooked (table 2; online supplementary table 2), although are not required for efficacy in MS—alemtuzumab, one of the most effective disease modifiers available, has no known effect on the BBB. Effects on leucocyte extravasation and BBB permeability have been most widely studied, with IFN-β, fingolimod and natalizumab all thought to act to increase BBB integrity. It is interesting that some therapies have presumably unanticipated effects at the BBB. For example, laquinimod, which is yet to be approved, has been shown to act directly on the endothelium to reduce expression of adhesion molecules ICAM-1 and ALCAM and decrease permeability through increased expression of AJ proteins. Some drugs also have actions on astrocytes, with laquinimod downregulating the astrocytic response with effects on MMP expression, and fingolimod inhibiting astrocytic ceramide production. In MS, levels of ceramides are particularly increased in astrocytic end-feet, and these ceramides have been shown to disrupt the BBB, showing a direct link between fingolimod action on astrocytes and BBB disruption. The efficacy of fingolimod in one EAE model has been shown to require S1P1 modulation in astrocytes.
Actions of disease-modifying multiple sclerosis (MS) drugs at the blood-brain barrier (BBB). Drug names were searched for in PubMed with the keywords ‘blood brain barrier’, ‘endothelial’, ‘endothelium’, ‘astrocyte’, ‘pericyte’ and ‘tight junction’. Results were reviewed and the most relevant associations are discussed. All references for this table are included in the online supplementary table 2
Cerebral hypoperfusion and tissue hypoxia are intrinsically linked with BBB disruption, so drugs that are used in the cardiovascular system more generally might provide a new avenue for treatment in MS. Aspirin has no positive effect in MS and may be associated with an increased risk of haemorrhage.58 Statins have been shown to have beneficial effects in EAE models but are associated with increased disease activity in RRMS when used as an add-on therapy with IFN-β, suggesting a potentially antagonistic effect between the two. In progressive MS, statins reduce cortical atrophy and show more promise.59 Inhibiting the angiotensin system might also be beneficial since it has also been associated with neuroinflammation. Understanding of disease aetiology and vascular changes in MS might further provide new specific targets and therapies for both relapsing-remitting and progressive disease.
Synthesis and conclusion
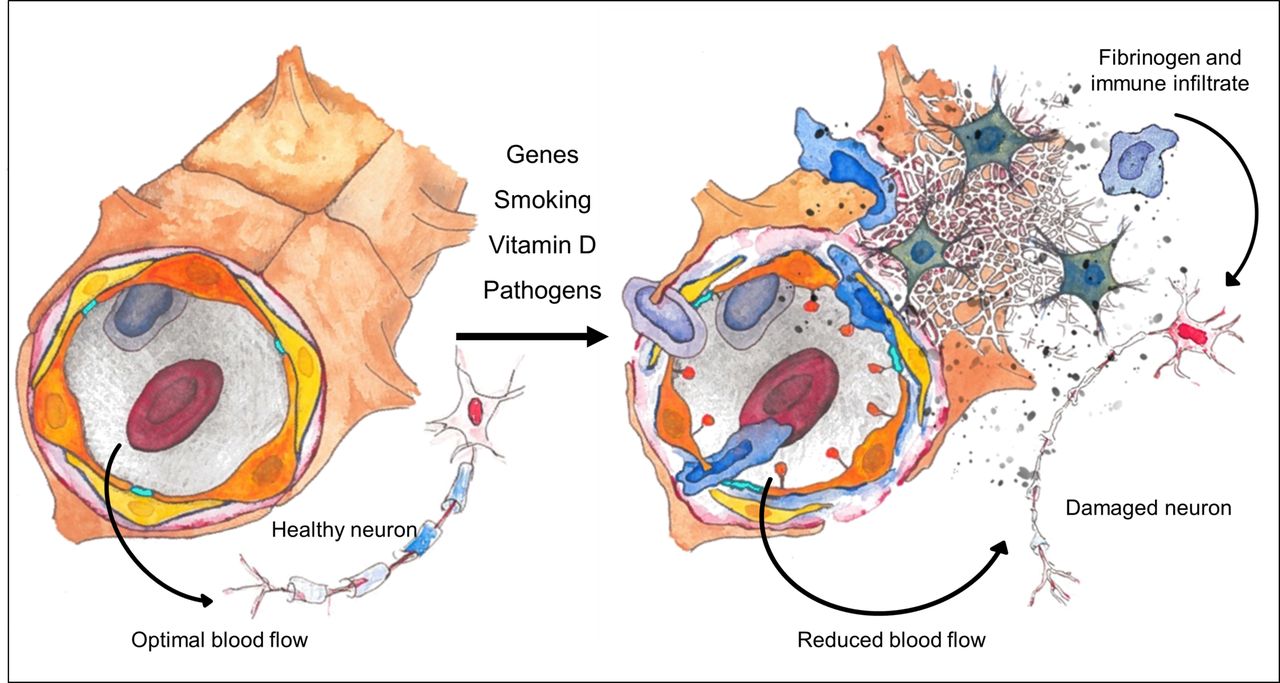
Our model proposes that known risk factors, as well as acting on immunomodulation, might also converge on the NVU (figure 3). A perfect storm of genetic and environmental factors could act together on the vasculature and the BBB to cause hypoxia, increased BBB permeability and altered immune trafficking. Within blood vessels, altered cerebral blood flow and BBB disruption appear to interact in a vicious cycle, which drives pathology. There are several avenues through which these early vascular changes might result in the formation of an MS lesion. A leaky BBB alone could allow exposure of naive leucocytes to immune privileged antigens and the production of an aberrant immune response. Fibrinogen deposition may also play an important role as it is able to directly activate microglia leading to neuronal damage60 and is able to induce inflammatory demyelination in the absence of T cells. Hypoxia caused by vascular changes or innate immune activation could directly cause oligodendrocyte loss, which in itself might enable activation of local microglia and inflammation. Enlarged VRSs caused by increased BBB permeability and inflammation might modulate antigen presentation and CNS antigen trafficking to cervical lymph nodes, allowing activation of adaptive immunity. Ultimately, an aberrant immune response against myelin, oligodendrocytes and neurons develops, leading to MS.
{kind=link}
{kind=link}
{kind=link}
Vascular pathology and multiple sclerosis (MS) pathogenesis. On the left, an intact neurovascular unit (NVU) and healthy neuron are depicted. The NVU contains endothelial cells (orange) with intervening tight junctions (TJs) (turquoise), pericytes (yellow) and an outer layer of astrocytic end-feet (brown) with the Virchow Robin space (VRS) lying between the pericytes and the astrocytic end-feet. On the right, a disrupted NVU and downstream consequences typically found in MS are depicted. Genetic and environmental risk factors associated with MS converge on the NVU to cause pathological changes, which ultimately lead to neuronal damage. These changes include endothelial activation (with expression of luminal adhesion molecules and chemokines), disruption of TJs and the basement membrane, engorgement of the VRS, retraction of astrocytic end-feet and vasoconstriction. These changes lead to extravasation of an immune infiltrate, deposition of fibrin(ogen) and reduction of oxygen delivery via hypoperfusion, which ultimately contribute to neuronal damage and death.
There is no single unifying theory of MS patho-aetiology. We have reviewed evidence showing that BBB disruption and vascular changes are a prominent and early feature of MS pathogenesis and may be influenced by genetic and environmental risk factors of MS. In synthesising this, we hypothesise that early disruption of the NVU could initiate a cascade of events which ultimately generates an immune response against the CNS and leads to MS. Importantly, this model does not go against current thinking of MS aetiology; we recognise that BBB dysfunction is necessary but is likely not sufficient for MS. While changes in the NVU may be a precipitating factor for the immune response against CNS components, a dysfunctional immune system is also required. However, regardless of the aetiological theory of MS one subscribes to, it is important to recognise that genetic and environmental risk factors, as well as current therapies, may also exert important effects on the vasculature and the BBB. Focus on the peripheral immune system alone may be limiting our understanding of the disease and the success of developing therapies.
Acknowledgments
The authors would like to acknowledge Professor Margaret Esiri for kindly offering to proof read the manuscript and Aimee Taylor, medical illustrator, for her assistance in creating Figure 3 entitled ‘Vascular pathology and MS pathogenesis’.
References
Footnotes
JIS and JSB contributed equally.
Contributors JIS and JSB are joint first authors. JIS, JSB and GCD conceived the idea for this review article. JIS and JSB carried out a review of literature as outlined in text and put together the figures. JIS, JSB and GCD together interpreted the literature and wrote the manuscript. All authors gave final approval for the submitted article and take responsibility for the accuracy and integrity of this work.
Competing interests GCD is supported by the NIHR Biomedical Research Centre (BRC), Oxford and has research funding from the Oxford BRC, MRC(UK) and Merck Serono. GCD has received travel expenses from Bay Schering, Biogen Idec, Genzyme, Merck Serono and Novartis, and honoraria as an invited speaker for Bayer Schering and Novartis.
Provenance and peer review Commissioned; externally peer reviewed.
Correction notice This article has been corrected sine it was published Online First. The Contributorship statement has been updated.