Article Text

Abstract
OBJECTIVES Myotonic dystrophy is a disease characterised by myotonia and muscle weakness. Psychiatric disorder and sleep problems have also been considered important features of the illness. This study investigated the extent to which apathy, major depression, and hypersomnolence were present. The objective was to clarify if the apathy reported anecdotally was a feature of CNS involvement or if this was attributable to major depression, hypersomnolence, or a consequence of chronic muscle weakness.
METHODS These features were studied in 36 adults with non-congenital myotonic dystrophy and 13 patients with Charcot-Marie-Tooth disease. By using patients with Charcot-Marie-Tooth disease as a comparison group the aim was to control for the disabling effects of having an inherited chronic neurological disease causing muscle weakness. Standardised assessment instruments were used wherever possible to facilitate comparison with other groups reported in the medical literature.
RESULTS There was no excess of major depression on cross sectional analysis in these patients with mild myotonic dystrophy. However, apathy was a prominent feature of myotonic dystrophy in comparison with a similarly disabled group of patients with Charcot-Marie-Tooth disease (clinician rated score; Mann Whitney U test, p=0.0005). Rates of hypersomnolence were greater in the myotonic dystrophy group, occurring in 39% of myotonic dystrophy patients, but there was no correlation with apathy.
CONCLUSION These data suggest that apathy and hypersomnia are independent and common features of myotonic dystrophy. Apathy cannot be accounted for by clinical depression or peripheral muscle weakness and is therefore likely to reflect CNS involvement. These features of the disease impair quality of life and may be treatable.
- myotonic dystrophy
- apathy
- major depression
- hypersomnia
Statistics from Altmetric.com
Myotonic dystrophy is the commonest form of adult muscular dystrophy, with a population incidence of 1:8000.1 This progressive multisystem disease is inherited in an autosomal dominant manner and is associated with an abnormal expansion of a (CTG)n repeat in the 3′ untranslated region of a protein kinase gene on chromosome 19. Normal chromosomes have 5–37 copies of the CTG repeat, whereas myotonic dystrophy is associated with 50 to 2000 repeats. The number of triplet repeats correlates broadly with the severity of the physical phenotype and with earlier age of onset.2 3 The cardinal features are myotonia or muscle weakness, which often occur in association with cataracts, frontal balding, and specific cardiac and gastrointestinal manifestations. Patients with the congenital and childhood onset are often severely affected and have more pronounced cognitive impairment.
The CNS is known to be affected. For example, Rosman and Kakulas4 commented on blunted, foreshortened, and flattened frontal lobes in three necropsy cases with known cognitive impairment. The presence of eosinophilic inclusion bodies in the dorsomedial nucleus of the thalamus5-7 and rod-like intracytoplasmic inclusions in the large neurons of the caudate have also been reported.8 Neuroimaging studies, summarised in Avrahami et al 9 have found small sella turcicae, overdevelopment of the frontal sinuses, hyperostosis of the calvarian bones, cerebral atrophy, hydrocephalus, and progressive ventricular dilatation. A CT study found hyperostosis and corresponding microcephaly in 71% and ventriculomegaly in 13% of patients.9 However, using MRI, ventriculomegaly was evident in 71%.10 Studies with MRI are conflicting with respect to the presence of white matter lesions.10-13 A PET study using F labelled 2-fluoro-2-deoxy-D-glucose showed that cortical glucose utilisation was reduced by 20% in myotonic dystrophy compared with controls.14 This concurs with single photon emission computerised tomography (SPECT) findings of decreased cerebral perfusion especially in the frontal and temporoparietal association cortex.13
Cognitive impairment in myotonic dystrophy is well recognised. In the more severely affected congenital group, Harper15 reported a mean IQ of 66 (SD 16). Although studies looking at the whole range (congenital, child, and adult onset cases) have shown a lower mean IQ score than normal controls (reviewed by Turnpenny et al 16), IQ has recently been shown not to be impaired in people with mild myotonic dystrophy.16 17However, studies which have examined cognitive function in more detail have highlighted difficulties with particular tasks such as attention,18 visual memory, visual construction, and perception13 as well as executive function (see discussion). It is likely that virtually all myotonic dystrophy studies performed before the mutation was identified in 1992 (reviewed by Wieringa1) were affected by ascertainment bias towards the more severe forms of the disease, as patients with minimal symptoms and equivocal disease status would have been excluded. It is also unclear if some congenital cases or very severely affected cases were primarily responsible for the excess of cases with low scores.
There have been anecdotal reports of apathy, severe moodiness and suspiciousness, and excessive daytime sleepiness occurring in association with myotonic dystrophy.2 3 19 Bird et al 18 and Palmer et al,20using personality inventories, concluded there was no one personality type associated with myotonic dystrophy. Apathy is consistently reported in the literature.3 19 21 Caughey and Myrianthopoulos22 reported “that affected individuals when just mildly incapacitated were often content to sit or lie idle for hours”. In a different context, apathy has been defined as “a lack of motivation not attributable to a diminished level of consciousness, cognitive impairment, or emotional distress”.23 However, apathy in myotonic dystrophy has not been clearly defined and its relation with excessive daytime sleepiness or depression has not been investigated. The reasons for excessive daytime sleepiness in myotonic dystrophy are unclear. Whereas some have argued that this is the result of sleep apnoea, recent data suggest that this symptom is usually caused by a dysfunction of central sleep regulation and is not a consequence of disturbed nocturnal breathing.24 It is also unclear whether apathy in myotonic dystrophy is part of a depressive syndrome. High levels of depressive symptoms have been described11 12 and Brumback et al 25 reported that all of their 15 patients with myotonic dystrophy met DSM III criteria for major depression. However, many of the depression scales used in these studies do not allow a formal diagnosis of major depression to be made and simply reflect a range of depressive symptoms.
To our knowledge, systematic investigation of apathy in myotonic dystrophy has not been undertaken. The aim of this study was to quantify the degree of apathy in people with myotonic dystrophy and to see if this was related to the extent of neuromuscular involvement, the presence of hypersomnolence, or to major depressive syndrome. We hypothesised that apathy may be an independent feature of myotonic dystrophy reflecting CNS involvement. To clarify this we studied those with milder disease to lessen the potential effects of severe weakness and disability. In addition, patients with Charcot-Marie-Tooth disease were included as a comparison group to control for muscle weakness, chronic disability and the presence of an inherited disease. Charcot-Marie-Tooth disease is a disease of the peripheral nervous system which, to the best of our knowledge, does not affect the CNS. In this paper we present the findings on apathy, psychopathology, and sleep in those with myotonic dystrophy compared with the Charcot-Marie-Tooth disease control group.
Methods
The study was undertaken in the East Anglian area served by the Regional Genetics Service. Approval was obtained for this study from the local area ethics committee.
SELECTION OF PATIENTS AND CONTROLS
Subjects were identified from the clinical genetics service rather than the neurological clinic so that patients with the full range of disability would be included. This clinical genetics department has made a concerted effort to trace families with myotonic dystrophy to offer molecular diagnosis. Ascertainment towards mild disease was considered preferable so that psychopathology, if present, would be less likely to be directly attributable to disabling disease.
All those between 18 and 70 years of age in the East Anglia area who had had a DNA diagnosis of myotonic dystrophy were initially considered for inclusion. Those with childhood onset, adult onset, or the late onset form of the disease were included but those with congenital myotonic dystrophy (with signs of the illness from birth) were not. Patients were also excluded if other neurological illnesses known to affect muscle or cognitive function were present. Permission was sought from the responsible clinical geneticist or genetic nurse specialist as well as from the patients’ general practitioner before seeing the subject. A further set of patients were excluded at the request of their clinicians. Reasons for exclusion were a recent termination of pregnancy, a recent family death, and a third trimester pregnancy. Two patients were excluded because of current adverse social circumstances and another at the request of a general practitioner (GP) because of postnatal depression. Subjects with Charcot-Marie-Tooth disease had all been seen in the department of clinical genetics and the diagnosis of Charcot-Marie-Tooth disease type1 had been made on the basis of clinical findings and nerve conduction studies which were typical, or by the presence of the characteristic chromosome 17 duplication.26
A total of 57 patients with myotonic dystrophy and 19 patients with Charcot-Marie-Tooth disease were contacted by letter; 21 and six respectively refused or did not reply. Thus 36 patients with myotonic dystrophy and a comparison group of 13 cases with CMT were investigated. Among the cases with myotonic dystrophy there were five sibling pairs, one family with three siblings and three parent-child pairs. There were two sibling pairs in the Charcot-Marie-Tooth disease sample.
Subjects were seen at home in nearly all cases (46 of 49). The researcher (JSR) could not be blinded to the diagnosis. Testing was completed in about four hours with appropriate breaks.
ASSESSMENT INSTRUMENTS
A structured questionnaire was administered to obtain the subject’s age, duration of illness, maternal or paternal inheritance of disease, and age of onset of the illness. The muscle weakness or myotonia or both as first noted by the patient defined the onset of the disease (as in Hunter et al 2). In addition, questions were asked to elicit other medical conditions, current medication, schooling, and employment record.
The Maudsley Hospital sleep questionnaire (J D Parkes, personal communication) was used to assess sleep patterns. This questionnaire elicits symptoms about the main sleep disorders and contains the Epworth sleepiness scale.27
The following psychopathology rating scales were used:
(1) Apathy evaluation scale28—the clinician rated and self rated versions of this scale were administered. This is an 18 item questionnaire in which there are four options per question on a continuum. The higher the score the greater the apathy. The validity and reliability (internal consistency, test-retest, and interrater) of these apathy scales have been established.28
(2)Fatigue questionnaire29—a 13 item graded and self rated questionnaire which is divided into physical and mental symptoms relating to fatigue was given.
(3) The schedule for affective disorder and schizophrenia-lifetime version (SADS-L) was used.30
General intelligence was rated using the National adult reading test.31
Muscle power was assessed using the MRC United Kingdom scale.32 There was no established test of functional disability available for patients with peripheral nerve or muscle weakness and therefore a scale was devised which assessed muscle functioning by asking about the ability of the patients to undertake day to day tasks or activities. The questionnaire covers if they were able to manage the following six tasks: walk up 15 stairs, ride a bicycle, open a jar easily, lift heavy parcels (a standard example of four full shopping bags was given), lift themselves out of the bath, and lift their head off the bed if they were lying flat on it (without turning on the side). This was then used to group patients into mild (one to two negative replies), moderate (three to four negative replies), and severe (five to six negative replies). The age of onset of the muscle weakness and or myotonia was obtained. This correlates broadly with triplet repeat size.2 33 and therefore provides an indirect measure of the degree of disability.
For genetic analysis, CTG triplet repeat sizes were determined using Southern analysis.33
Statistical analysis was performed using the SPSS-PC software package (SPSS Inc, Chicago, Ill). Much of the data were ordinal, so non-parametric statistics were used.
Results
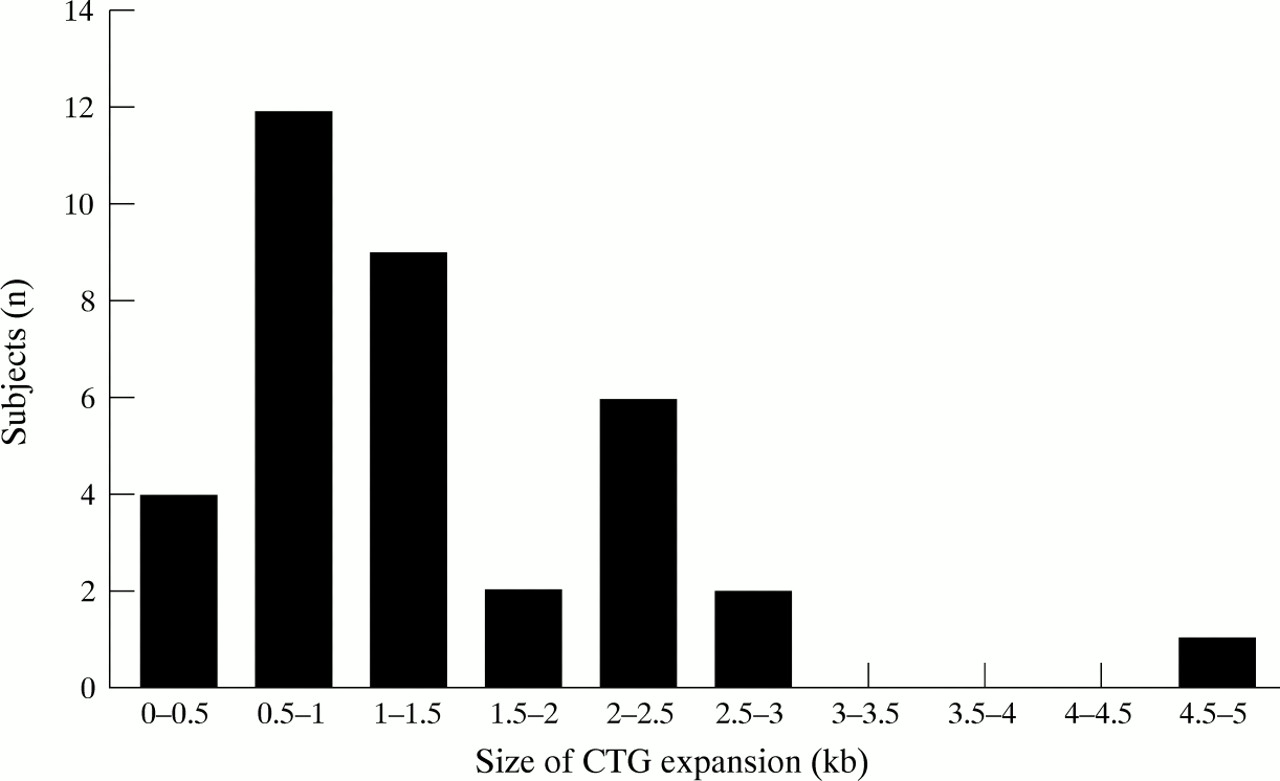
Table 1 shows the demographic characteristics of the myotonic dystrophy and Charcot-Marie-Tooth disease groups. These groups had similar mean ages, male:female ratios, years of education, and IQ levels. Other cognitive data from the patients with myotonic dystrophy have been reported separately.17 In the myotonic dystrophy group, 22 patients had mild disability (61%), seven (19%) moderate, and seven (19%) severe. In the Charcot-Marie-Tooth disease group five (39%) patients had mild disability, seven (54%) moderate, and one (8%) severe. The degree of disability in the myotonic dystrophy and Charcot-Marie-Tooth disease groups were not significantly different. CTG expansions in the myotonic dystrophy cases ranged from 0.3–4.6 kb (fig 1). The age of onset of the patients with myotonic dystrophy was as follows: no patients with age of onset under 10 years, five patients (14%) between the ages of 10–17, 20 patients (56%) between the ages of 18–39, 10 patients (28%) aged 40 and above; one subject had no clinical manifestations but had DNA confirmation of myotonic dystrophy.
Demographics of myotonic dystrophy (MD) and Charcot-Marie-Tooth (CMT) groups

CTG repeat sequence expansion (in kilobases (kb)) in patients with myotonic dystrophy. The total number of patients with myotonic dystrophy is 36.
APATHY
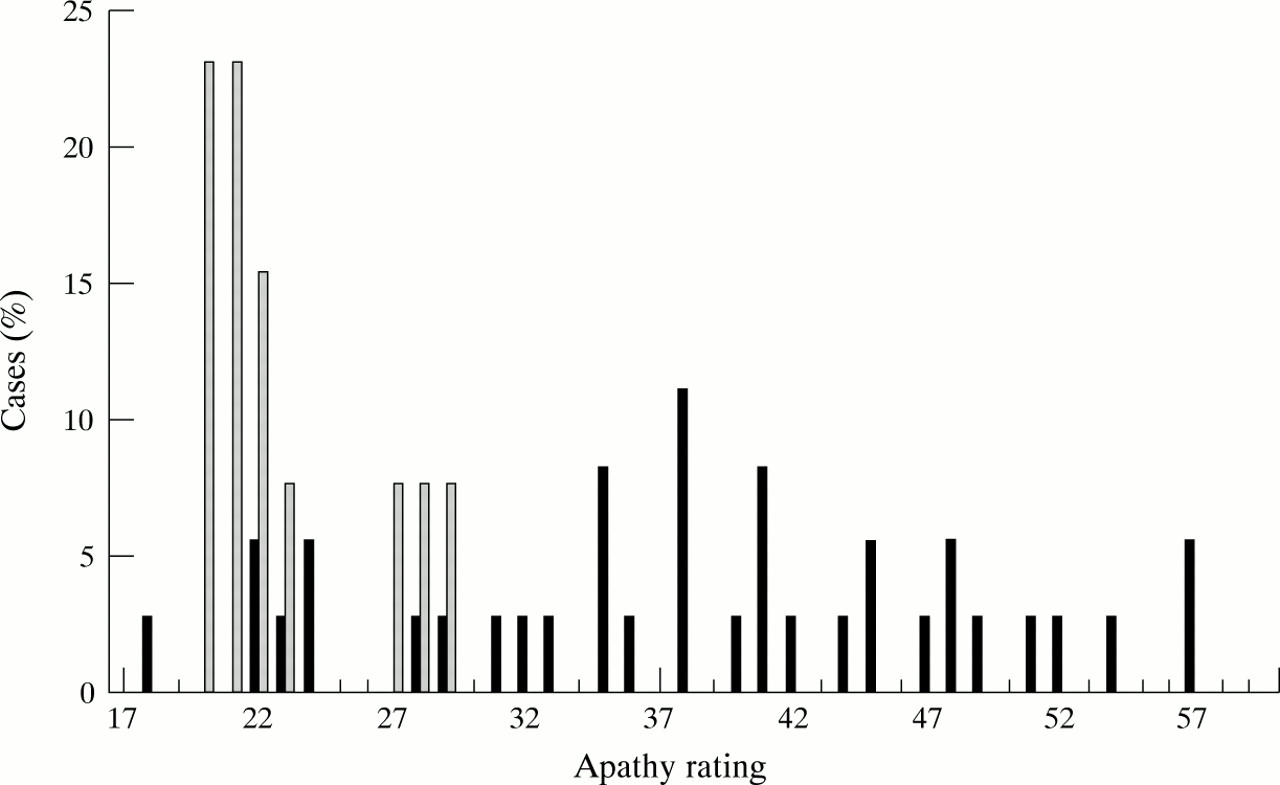
Significantly higher apathy scores were found in the myotonic dystrophy group than in the Charcot-Marie-Tooth disease group. Even when the two patients with current diagnoses of depression (and high apathy scores) were excluded from these calculations, the difference in apathy scores remained significant (Mann-WhitneyU test, p=0.0001—self rated and p=0.0005—clinician rated, fig 2). There was a significant correlation between clinician and self rated apathy scores (Pearson correlation coefficientr=0.796, p=0.001) and therefore further correlations were only performed using clinician rated scores. No correlations were found between apathy and duration of illness, age, CTG repeat length, or severity of disability.
{kind=link}
{kind=link}
Clinician rated version of the apathy evaluation scale28 using patients with myotonic dystrophy (black bars) and patients with Charcot-Marie-Tooth disease (striped bars).
FATIGUE
Mental and physical fatigue were not significantly different in the myotonic dystrophy v Charcot-Marie-Tooth disease cases (p=0.145 and 0.52 respectively; Mann-Whitney U test). Apathy did not correlate with either mental or physical fatigue scores suggesting that “apathy” is a separate entity from fatigue.
HYPERSOMNIA
Fourteen patients with myotonic dystrophy (39%) met minimal diagnostic criteria for idiopathic “hypersomnolence”.34 However, in the presence of myotonic dystrophy, the hypersomnolence cannot be considered truly idiopathic. In addition, two patients with myotonic dystrophy had symptoms of sleep apnoea as well as being hypersomnolent. One has had this diagnosis subsequently confirmed by sleep studies and is receiving treatment. The other patient refused investigation. None of the patients with Charcot-Marie-Tooth disease had hypersomnia although five complained of secondary insomnia (secondary to cramps in their legs). The myotonic dystrophy group (excluding two patients with sleep apnoea and two depressed patients) were compared with the Charcot-Marie-Tooth disease group (from which we excluded the cases with secondary insomnia). Significantly more patients with myotonic dystrophy had hypersomnia (Fisher’s exact test, p=0.03). In the hypersomnolent group there was only one pair of siblings; the rest were unrelated.
Functional disability was more pronounced in the hypersomnolent compared with the non-hypersomnolent cases with myotonic dystrophy (Mann Whitney U test, two tailed, p=0.0027). However, the presence of hypersomnolence in myotonic dystrophy did not correlate significantly with apathy scores, duration of illness, age, or CTG repeat length (Mann-Whitney U tests were used in all comparisons).
DEPRESSION
A current diagnosis of major depression was found in two of 36 (5.6%) patients with myotonic dystrophy and in none of the patients with Charcot-Marie-Tooth disease (difference not significant, table 2). The current point prevalence for major depression in cases of myotonic dystrophy is similar to that in the large epidemiological survey of Weissman and Myers,35 who also used the SADS-L (χ2=0.13, p=0.72).
Current and lifetime depression rates of patients with myotonic dystrophy (MD) and Charcot-Marie-Tooth (CMT) disease using SADS-L
Fifteen of 36 (42%) patients with myotonic dystrophy and four of 13 (31%) patients with Charcot-Marie-Tooth disease met the research diagnostic criteria (RDS)30 for “probable or definite lifetime history of major depression” (there was no significant difference between the two groups, table 2). Lifetime depression ratings in myotonic dystrophy showed a trend towards being raised in the myotonic dystrophy group compared with the data of Weissman and Myers.35 When our data were subdivided by sex, women with myotonic dystrophy showed increased lifetime depression scores compared with these epidemiological data. This trend was not found in men (table2). Depression was often temporally related to divorce or separation, and less commonly followed diagnosis or worsening of the illness. In four of 15 patients with myotonic dystrophy the onset of depression predated the onset of the myotonia or muscle weakness and in only four patients was there a recurrent history of major depressive episodes. There was no apparent excess of the following psychopathologies in myotonic dystrophy: alcohol misuse (two of 36), generalised anxiety (two of 36), labile personality disorder (one of 36), intermittent depressive disorder (one of 36), and minor depressive episode (two of 36). Some patients had more than one diagnosis during their lifetime using RDC rules.30
Discussion
Myotonic dystrophy is generally thought of as a neurological disease with the main emphasis placed on the characteristic progressive myotonia and muscle weakness. In this study we have focused on the psychopathological features and abnormal sleep patterns associated with myotonic dystrophy. We think that these aspects of myotonic dystrophy are important for three main reasons. Firstly, if they are found to be a significant part of the phenotype then this knowledge will help people with myotonic dystrophy and their families to understand their problems better. Secondly, some of these associated problems may be amenable to treatment and thus lead to an improved quality of life. Thirdly, if a specific psychopathology can be shown to be the direct consequence of the myotonic dystrophy mutation (as opposed to being, for example, secondary to muscle weakness), there is the potential for investigating the molecular biological basis for that psychopathology. Thus we think that it is important to investigate and quantify psychopathology in genetic diseases in which it has been anecdotally reported. Our data suggest that apathy and hypersomnolence are common and independent features of myotonic dystrophy.
Our study design was constrained by practical considerations. Firstly, the investigator seeing the patients was not blinded to their diagnosis. This is not an easy problem to overcome, as many patients with myotonic dystrophy have characteristic facies. However, the SADS-L30 is a structured interview, which will reduce interviewer bias, and apathy and hypersomnolence were assessed with questionnaires completed by the study patients. Secondly, 10 of the 36 patients with myotonic dystrophy were first degree relatives and psychopathology may be familial. However, this does not alter our findings of the relation between having myotonic dystrophy and the presence of apathy and hypersomnolence. It would only give rise to a spurious interpretation if the increased apathy and hypersomnolence were due to the inheritance of a gene closely linked to the myotonic dystrophy protein kinase gene and cosegregating with it, or if these features were strongly determined by common familial environmental factors. Both of these seem unlikely. Thirdly, although this is one of the largest series of patients with myotonic dystrophy examined for psychopathology, the sample size is not large enough to confidently exclude effects where no significant correlations or associations were detected. In addition, we have specifically excluded patients with congenital disease, who are more likely to have larger CTG expansions. As the points at the extremes have greater weight in correlation analyses, the paucity of severe cases may have diminished our ability to relate psychopathology to severity of disease or CTG repeat length.
The low current prevalence rates of depression in patients with myotonic dystrophy (two of 36) are very different to those found by Brumback et al. 25 who found current depression in 100% of their patients. This may be accounted for in part by the milder degree of functional disability in our patients. The difference in lifetime depression rates in patients with myotonic dystrophy versus the epidemiological control group were only of borderline significance in women. This sample may not have shown significance for men because of the fewer men seen. Other than the one patient with postnatal depression who was excluded at the request of her clinician, there seemed to be no bias against selection of patients with depression.
Hypersomnia is a common and disabling symptom in myotonic dystrophy. About one third of patients with myotonic dystrophy have sleep disorders.36 In the general population 3.7%-4.2% describe hypersomnolence.37 Thirty nine per cent of our patients met criteria for “idiopathic hypersomnolence” and a further 6% had both sleep apnoea and hypersomnolence. A recent study on those with myotonic dystrophy referred to a sleep disorders clinic reported that 14 of 22 (63.6%) were hypersomnolent but did not fulfil criteria for sleep apnoea (although both diagnoses were present in a further three patients).24 This study concludes that the hypersomnolence seen was not explicable purely in terms of sleep apnoea alone but is due to a disturbance in central sleep regulation. Hypersomnolence is likely to be an independent feature to apathy in myotonic dystrophy, as the hypersomnolent patients did not have greater levels of apathy than the non-hypersomnolent group. In five of 13 cases, hypersomnolence seemed to predate the onset of muscle involvement and in another five cases occurred at the same time as these peripheral features, suggesting that this symptom is unlikely to be secondary to general or respiratory muscle weakness (which tends to occur late in the illness). There was greater functional disability in the hypersomnolent group, so hypersomnolence may follow the course of the illness more closely in the later phase, or may be independently associated with the extent of the disability.
Subjects with myotonic dystrophy show increased apathy compared both to patients with Charcot-Marie-Tooth disease and literature controls. The mean (SD) of the clinician rated apathy score for patients with myotonic dystrophy was 38.4 (10.4) and for the patients with Charcot-Marie-Tooth disease 23.7 (4.4). The mean scores (SD) reported previously in the following groups are: normal elderly sample 26 (6.2), left hemispheric stroke 31.9 (9.6), right hemispheric stroke 34.7 (7.3), Alzheimer’s disease 44.4 (11.1), and major depression 40.5 (9.7) (one of the sets of clinician rated scores from Marin et al.28 )
Our ascertainment of patients on the basis of DNA records has ensured the inclusion of patients with milder symptomatology, who may previously have gone unnoticed. This has, in part, allowed us to conclude that apathy is not simply a feature of the severely affected patients with myotonic dystrophy. As the Charcot-Marie-Tooth disease group did not have apathy, we concluded that apathy is not a general secondary psychological reaction related to having a chronic genetic disease causing muscle weakness. The very limited number of patients with myotonic dystrophy showing major depression when interviewed, mitigate against explaining apathy simply as part of a depressive syndrome.
The symptom of apathy is seen commonly in patients with a frontal lobe syndrome and these patients perform poorly on executive function tasks. In myotonic dystrophy there are conflicting reports about frontal lobe tasks being affected. Whereas some11 12 38 39 found frontal lobe tasks such as the Wisconsin card sorting test or the verbal fluency test to be affected, others20 40 report differences of borderline significance or no deficits. This discrepancy may be due to the exclusion of congenital cases.20 This is particularly relevant as executive function tests are particularly sensitive to low intelligence. We have compared patients with myotonic dystrophy with normal age and IQ matched controls17 on three tests of executive function—namely, a verbal fluency task, the modified Wisconsin card sorting test,41 and the cognitive estimates test.42 Patients with myotonic dystrophy were not significantly affected on the first but results reached borderline significance (p=0.053 and p=0.047) on the second two tests. These findings, combined with the apathy we documented, are consistent with frontal lobe involvement. In addition, this hypothesis is supported by pathological findings involving the frontal lobes4 and frontal hypoperfusion on SPECT.13
Apathy has been found and quantified in other neuropsychiatric disorders—for example, in patients with Parkinson’s disease,43 cerebrovascular disease,28 44 and Alzheimer’s disease.28 Although little is known about the neurochemistry of myotonic dystrophy, it is interesting to consider whether there may be common mechanisms underlying apathy in myotonic dystrophy and other disorders. Marin23 suggested that a common neurochemical basis underlies some disorders presenting with apathy. For instance, a functional deficiency of dopaminergic systems may explain the apathy seen in neuroleptic induced akinesia, the negative symptoms in schizophrenia, and the apathy in subcortical dementias such as Parkinson’s disease and frontal lobe syndromes.23 However, he has argued that this simple model is unlikely to explain apathy in other disorders, such as Alzheimer’s disease, in which there is damage to the hippocampus, amygdala, and contiguous structures. The role of the amygdala and closely related temporal-diencephalic subsystems in apathy is suggested by the Kluver-Bucy syndrome in monkeys.45
In conclusion, we think that apathy and hypersomnia may be CNS features of the myotonic dystrophy mutation. Apathy in these patients did not correlate with hypersomnolence or functional disability. In addition, most patients did not have clinical depression when interviewed and therefore the high apathy levels in the group as a whole cannot be explained in these terms. Although myotonic dystrophy is considered primarily as a neurological disease, we think that apathy and hypersomnia are integral parts of the disease and reflect CNS involvement. These features of the illness impair quality of life and should be considered as they may be treatable.
Acknowledgments
This research project was undertaken during a research post on the Cambridge Psychiatric Training Scheme. Funding was received from Lifespan Community NHS Trust and Merck-Lipha Research prize for Psychiatry. We acknowledge all the participating patients and their general practitioners, the generous assistance of the Medical Genetics Department, Addenbrooke’s Hospital and Mr D Dow, Dr C Walsh, Professor RS Marin, Dr J Sussman, and Dr GE Berrios. Special thanks to Mrs R Patterson for secretarial assistance.