Article Text

Abstract
OBJECTIVE To analyse the natural history and survival of corticobasal degeneration by investigating the clinical features of 14 cases confirmed by postmortem examination.
METHODS Patients with definite corticobasal degeneration were selected from the research and clinical files of seven tertiary medical centres in Austria, the United Kingdom, and the United States. Clinical features were analysed in detail.
RESULTS The sample consisted of eight female and six male patients; mean age at symptom onset was 63 (SD 7.7) years, and mean disease duration was 7.9 (SD 2.6) years. The most commonly reported symptom at onset included asymmetric limb clumsiness with or without rigidity (50%) or tremor (21%). At the first neurological visit, on average 3.0 (SD 1.9) years after symptom onset, the most often encountered extrapyramidal features included unilateral limb rigidity (79%) or bradykinesia (71%), postural imbalance (45%), and unilateral limb dystonia (43%). Ideomotor apraxia (64%), and to a lesser extent cortical dementia (36%), were the most common cortical signs present at the first visit. During the course of the disease, virtually all patients developed asymmetric or unilateral akinetic rigid parkinsonism and a gait disorder. No patient had a dramatic response to levodopa therapy. Median survival time after onset of symptoms was 7.9 (SD 0.7) (range, 2.5–12.5) years, and, after the first clinic visit, 4.9 (SD 0.7) (range, 0.8–10) years. Early bilateral bradykinesia, frontal syndrome, or two out of tremor, rigidity, and bradykinesia, predicted a shorter survival.
CONCLUSION The results confirm that unilateral parkinsonism unresponsive to levodopa and limb ideomotor apraxia are the clinical hallmarks of corticobasal degeneration, and only a minority of patients with corticobasal degeneration present with dementia. The study also suggests that a focal cognitive and extrapyramidal motor syndrome is indicative of corticobasal degeneration. Survival in corticobasal degeneration was shortened by the early presence of (more) widespread parkinsonian features or frontal lobe syndrome.
- corticobasal degeneration
- atypical parkinsonism
- survival
- natural history
- clinicopathological study
Statistics from Altmetric.com
Corticobasal degeneration is increasingly recognised as a distinctive neuronal multisystem degeneration presenting as an atypical parkinsonian syndrome.1 2 Although several characteristic clinical features have been proposed, such as unilateral levodopa unreponsive akinesia and rigidity, dystonia, or myoclonus, as well as cortical signs such as ideomotor apraxia and cortical sensory loss,1 3 many patients are misdiagnosed during life as having Parkinson’s disease, progressive supranuclear palsy, multiple system atrophy, or some other neurodegenerative disorder. In a recent study on clinical diagnostic accuracy in cases with pathologically confirmed corticobasal degeneration, sensitivity for the diagnosis was low at the first visit (35%), but specificity was near perfect (99.6%).4 Moreover, there was only a modest increase in sensitivity at the last visit. Furthermore, a recent clinicopathological study disclosed a striking clinical heterogeneity when analysing definite corticobasal degeneration cases with associated neuropathological disorders.5
To precisely delineate the range of clinical features in this disorder, we characterised 14 cases of corticobasal degeneration confirmed at postmortem examination. In addition, we analysed the natural history and clinical predictors of survival in these patients with definite corticobasal degeneration acquired from seven brain banks specialising in the study of parkinsonian syndromes.
Methods
Patients were selected from the research and clinical neuropathological files of seven medical centres in three countries (Austria, the United Kingdom, and the United States). Ten patients were previously included in a study on the accuracy of the clinical diagnosis of corticobasal degeneration4; the other four were added subsequently. Three patients were prospectively followed up until death by one of the coauthors (JJ). Most patients were assessed in movement disorders clinics; however, several general neurologists performed some of the first visits. All cases met the recently published National Institute of Neurological Disorders and Stroke (NINDS) preliminary neuropathological criteria for corticobasal degeneration.6 The hallmarks of corticobasal degeneration pathology, according to the NINDS criteria, include frontoparietal atrophy with severe cortical neuronal loss and intense astrogliosis as well as swollen and achromatic neurons (without Pick bodies) in the affected cortex. Several cytoskeletal abnormalities were also present in various combinations including neuropil threads, neurofibrillary tangles, and basophilic inclusions (the last in the substantia nigra and other basal ganglia areas). Tau positive astrocytic inclusions forming cortical astrocytic plaques were also found. At present such astrocytic plaques are considered highly characteristic of corticobasal degeneration. Other disorders mimicking some of the features associated with corticobasal degeneration such as multiple infarcts, profound diffuse atrophy, Lewy bodies, changes diagnostic of Alzheimer’s disease, oligodendroglial argyrophilic inclusions, Pick bodies, and prion P positive amyloid plaques must be excluded.
Patients’ records were abstracted on standardised forms by seven of us (AM, KJ, KRC, RKBP, LD, IL, CAM) who followed strict instructions. Because the data were retrospectively collected in 11 of the 14 patients, a feature that was not specifically recorded was coded as absent if the neurological examination was stated to be within normal limits. If the examination disclosed abnormalities without recording a particular feature, this was coded as missing.
Survival was calculated by the Kaplan-Meier analysis. Variables are expressed as mean (SD). Student’s t tests for independent groups as well as Pearson’s correlation analysis were applied as appropriate.
Results
DEMOGRAPHICS
Table 1 shows the main demographic characteristics of the 14 patients (eight women and six men). Mean age at onset of corticobasal degeneration symptoms was 63 (7.7) years; mean duration from symptom onset to first neurological visit was 35.5 (27.2) months; and mean age at death was 71.7 (7.1) years. None of the patients had a familial parkinsonian disorder.
Demographic characteristics of patients with corticobasal degeneration confirmed at postmortem examination
INITIAL SYMPTOMS
Table 2 shows the initial corticobasal degeneration symptoms. Limb clumsiness was present in 50% of the patients. Gait disturbance with postural instability was noted by 36% of patients with falls present during the first year of symptom onset in 21%. Sensory dysfunction of one limb (for example, paraesthesiae, pain) was noted by 39% of the patients and led to (futile) surgical decompression of peripheral nerves or roots in some of them. Cognitive (memory deficits) or behavioural changes (frontal lobe type symptomatology such as apathy, irritability, disinhibition) occurred initially in 21% of the patients, and speech problems in 14%.
Initial symptoms in patients with corticobasal degeneration confirmed at postmortem examination
SYMPTOMS AT FIRST VISIT
The first clinic visit (table 3) occurred on average 3.0 (1.9) years after onset of symptoms. At the first visit, eight (57%) of the patients had some degree of unilateral or grossly asymmetric akinesia and rigidity and two other (14%) patients had bilateral (mildly or moderately asymmetric) parkinsonism. Irregular tremor, present at rest, was documented in three (21%) patients; transient pill rolling tremor was reported by a general neurologist in one (7%) patient. Three (25%) patients had focal myoclonus (one had an irregular resting tremor). Unilateral limb dystonia was present in six (43%) patients. Laterocollis to the left was present in one patient. Gait disorders were almost always associated with postural instability and present on first neurological examination in six (43%) patients (small stepped (n=2), bradykinetic (n=4), broad based (n=3)); two patients required assistance when walking, and one was wheelchair bound. Speech was slow in five (36%) patients, slurred in four (29%), dysphonic in two (14%), and unintelligible in one (7%). Voluntary vertical gaze was impaired in five (36%) patients, two of whom (14%) also exhibited an abnormal horizontal gaze. Ideomotor apraxia (impaired mimicking gestures and hand postures) was present in seven (64%) patients. Four (29%) patients developed an early alien hand phenomenon (complex unintentional movements of one limb interfering with normal motor tasks). Three of these four patients had concomitant ideomotor apraxia, or dystonia, or both; one had cortical sensory loss. Extensor plantar responses were present in three (21%) of the patients. Cerebellar ataxia was recorded in four (31%) patients. Nine (64%) patients were not treated with levodopa at the time of the first neurological visit. Three of four patients treated with levodopa did not benefit from treatment; only one patient had a moderate (30–50%) response of unknown duration.
Features of patients with corticobasal degeneration confirmed at postmortem examination
SYMPTOMS AT LAST VISIT
The last clinical visit occurred a mean of 6.1(2.0) years after symptom onset. In general, symptoms and signs apparent early in the course of corticobasal degeneration steadily progressed up to the last visit. In most cases, bilateral but asymmetric bradykinesia and rigidity dominated the extrapyramidal syndrome. In 46% of the patients, there was unilateral limb dystonia; only 23% of the patients had superimposed focal myoclonus. Three (21%) patients had irregular tremor present at rest, and one also had focal myoclonus. Axial rigidity was present in 67% of the patients; however, limb rigidity was equally (45%) or more severe (22%) in all of them.

{kind=link}
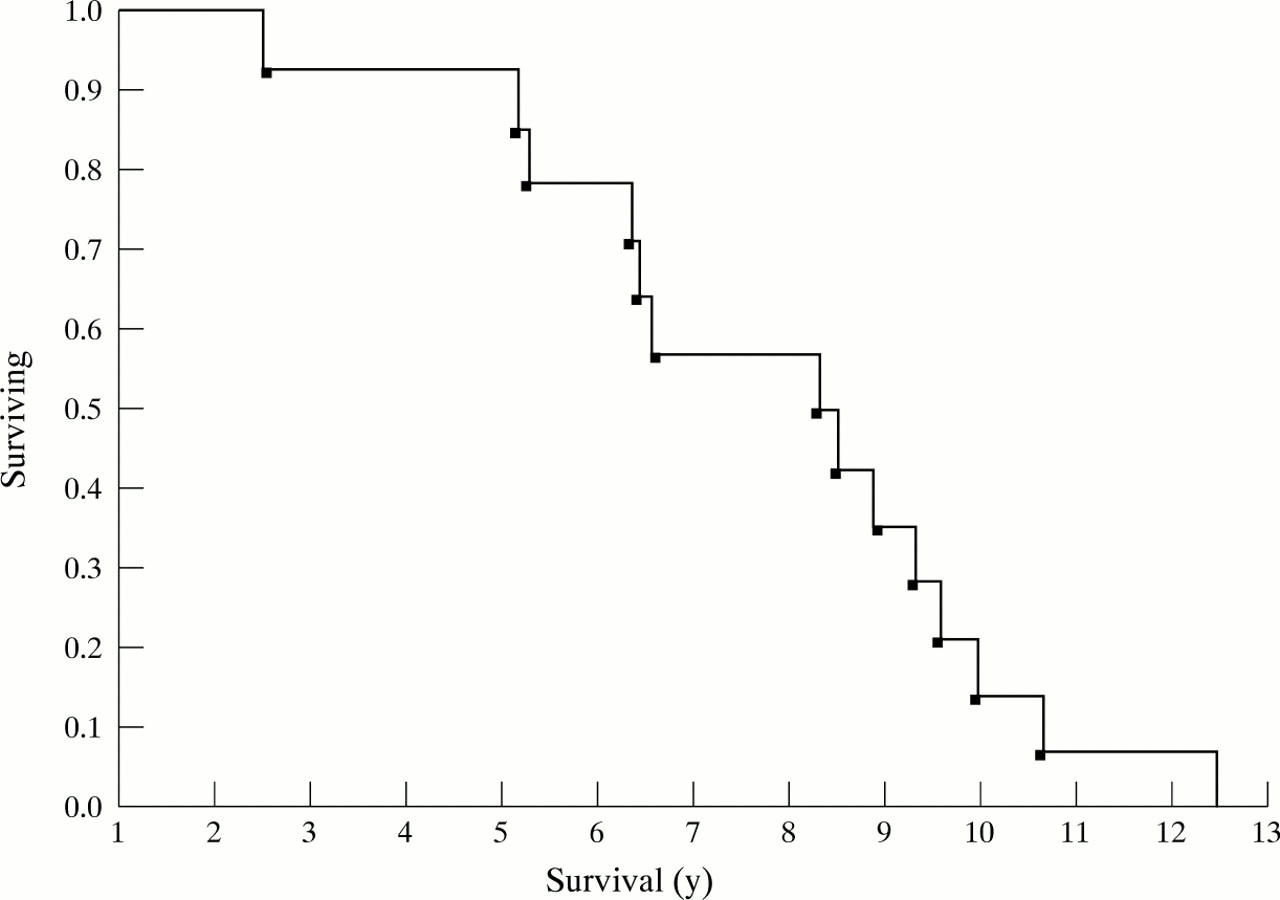
Kaplan-Meier table analysis of patients with corticobasal degeneration after onset of symptoms; the y axis refers to proportion of patients who are alive (surviving) at a given time point. The x axis is survival in years.
All patients developed an unstable and bradykinetic gait; 43% were wheelchair bound, and 14% required assistance to walk. Speech was almost always abnormal (93%): it was variably described as slurred (n=9), slow (n=9), dysphonic (n=5), mute (n=5), aphonic (n=4), unintelligible (n=4), echolalic (n=2), or palilalic (n=1). Ideomotor apraxia occurred in 75% of the patients; alien limb phenomenon was present in 46%. Aphasia was described in five (36%) patients: it was expressive motor type in two and mixed in three patients. Interestingly, the patients who developed aphasia had the onset of their motor symptoms on the right (three patients) or both limbs (two patients: one symmetric and one with asymmetric motor onset right>left), but never on the left. Four of the aphasic patients were right handed; handedness was not recorded in the remaining patient. Seven (58%) patients developed frontal lobe type symptoms such as apathy, irritability, or disinhibition, often associated with signs of frontal lobe release. Few patients presented pyramidal signs such as extensor plantar response (n=5) or hyperreflexia (n=7). True cerebellar signs were reported in two patients. Four patients received levodopa at the time of the last neurological visit with poor or no benefit.
SURVIVAL
Median survival from onset of corticobasal degeneration was 7.9 (0.7) (range, 2.5 -12.5) years (figure), and after the first clinical visit, it was 4.9 (0.7) (range, 0.8–10) years. Mean survival of patients with bilateral bradykinesia,or frontal syndrome, or both at the first visit was shorter (5.9 (3.4) years) than those without (11 years (0.7); p<0.05). Similarly, patients who exhibited two of three extrapyramidal features (tremor, rigidity, or bradykinesia) at the first examination had a shorter survival (7 (0.7) years) than those who did not (10.7 (1) years, p<0.01). These features were unrelated to duration of symptoms until the first visit. Neither age at onset nor early onset of dysphagia, dementia, incontinence, falls, gait disturbances, aphasia, ideomotor apraxia, pyramidal signs, dystonia, or vertical supranuclear palsy, had any effect on prognosis or survival.
CLINICAL DIAGNOSIS AND CAUSE OF DEATH
The primary neurologists made a diagnosis of corticobasal degeneration in five (36%) of the patients at the first visit. Of the remaining patients, three (21%) were undiagnosed; two (14%) were diagnosed as multiple system atrophy; and one (7%) patient each was diagnosed as atypical progressive supranuclear palsy, Pick’s disease, Alzheimer’s disease or symptomatic (for example, parkinsonism). At the last visit, a diagnosis of corticobasal degeneration was made in 50% of the patients; 14% each were either undiagnosed or thought to be symptomatic parkinsonism; and 7% each were diagnosed as atypical progressive supranuclear palsy, diffuse Lewy body disease, or other disease. Bronchopneumonia represented the cause of death in all patients with information available on the general postmortem findings.
Discussion
To our knowledge the present study is the first analysis of survival in corticobasal degeneration. Furthermore, the series of 14 pathologically established corticobasal degeneration cases represents the largest cohort reported to date. Our clinical data confirm that unilateral parkinsonism unresponsive to levodopa and limb ideomotor apraxia are the clinical hallmarks of corticobasal degeneration. A minority of corticobasal degeneration patients may present with early dementia or never present cortical features. Poor survival in our patients with corticobasal degeneration was predicted by the early presence of severe or bilateral parkinsonian features or frontal lobe syndrome. Our patients with corticobasal degeneration usually presented in the seventh decade and never before the age of 45. The most common initial manifestation was limb clumsiness, reported by half of the patients at the first visit. Other initial symptoms included gait disorder (36%) with falls (21%); unilateral painful paraesthesiae (29%); frontal lobe dementia symptoms (dementia associated with disinhibition) (21%); and dysarthria (14%). Similar presentations were previously described by Rinne et al in a large series of partly pathologically confirmed cases (table 3).1
At the first neurological visit, on average 3.0 years after onset of symptoms, the most common features included unilateral or asymmetric limb bradykinesia or rigidity (78%) as well as ideomotor apraxia (64%). However, only five (36%) patients were diagnosed as having corticobasal degeneration. The remaining patients were not diagnosed or thought to have other neurodegenerative disorders or symptomatic parkinsonian syndromes. The poor diagnostic accuracy may be partly related to neurologists’ limited awareness of the clinical range associated with corticobasal degeneration. However, other distinctive features of corticobasal degeneration such as alien limb syndrome, focal myoclonus, or dystonia, were only present in a few patients. In addition, levodopa treatment was not given in most cases; indeed, most neurologists considered that levodopa therapy was not useful. However, as parkinsonism associated with corticobasal degeneration is usually unresponsive to levodopa,1 antiparkinsonian therapy may facilitate an earlier diagnosis in some cases. Onset of motor symptoms in most of our patients was in the right extremities. Interestingly, those patients with onset of right motor symptoms developed aphasia or ideomotor apraxia, whereas one of the two patients with onset of motor symptoms on the left developed left sided visual neglect. These two right handed patients also developed constructive apraxia, but, probably because of bilateral involvement, also exhibited ideomotor apraxia. Thus in the absence of vascular events, the presentation of both ideomotor apraxia and left visual or sensory neglect should raise the suspicion of corticobasal degeneration. It is possible that diagnostic accuracy may increase if neurologists search for focal cognitive features such as neglect, aphasia, or ideomotor apraxia, and the alien hand phenomenon7 when they examine patients with asymmetric parkinsonism unresponsive to levodopa. Moreover, even if two thirds of the patients eventually developed ideomotor apraxia, this figure is probably an underestimate as neurologists do not always look for this feature.
Three patients experienced memory problems at the onset of their illness; other features suggestive of cortical dementia such as aphasia (n=2), ideomotor apraxia (n=2), or frontal lobe behaviour (n=2) were present at their first neurological visit. Thus as previously suggested,8 corticobasal degeneration should be considered in the differential diagnosis of dementia (and language disorders) particularly when accompanied by frontal lobe symptomatology, early speech alterations, or parkinsonism.
Several other features noted at the first visit deserve comment. A rest tremor was found in 29% of patients. In one patient, transient pill rolling tremor present at rest was described by the neurologist, suggesting that rest tremor (unrelated to superimposed myoclonus) may be present in a minority of patients with corticobasal degeneration. Differentiation from Parkinson’s disease may be difficult in this situation.
Axial symptoms, such as postural instability with or without falls and dysarthria, occurred in 29%-45% of the patients at first visit. These symptoms, together with vertical gaze palsy and frontal lobe release signs, may lead to an erroneous diagnosis of progressive supranuclear palsy.9 The gross asymmetry of parkinsonism and the presence of ideomotor apraxia should, however, caution against such misdiagnosis, although a minority of patients with progressive supranuclear palsy may develop mild ideomotor apraxia.10Furthermore, most patients with vertical gaze palsy also had equally severe horizontal gaze palsy, unlike the disproportionate involvement of vertical gaze in progressive supranuclear palsy .
At the last visit, grossly asymmetric bradykinesia and rigidity, gait disorder, and dysarthria had progressed unremittingly and were present in most or all patients. Βy contrast, cortical signs such as apraxia or frontal lobe release signs were absent in up to 40% of the patients. Furthermore, supposedly characteristic features such as asymmetric fixed dystonia, alien limb syndrome, or myoclonus were also absent in many of our patients.
Average disease duration (7.9 years) was slightly longer than in a recently reported series.1 Similar survival periods have been reported for other atypical parkinsonian syndromes such as multiple system atrophy and progressive supranuclear palsy.11-14 Among clinical features present at the first visit, only parkinsonian signs and frontal lobe features predicted a shorter survival, by contrast with dysphagia or gait disturbance. These features were unrelated to duration of symptoms until the first visit, and whether they represent a more aggressive form of the disease manifested as early bilateral involvement or are a statistical artifact, need further investigation. Other survival studies of multiple system atrophy or progressive supranuclear palsy patients also indicate that progressive parkinsonism unresponsive to levodopa determines survival in these related disorders.13 14 Most of our patients with corticobasal degeneration died from pneumonia as a result of dysphagia and immobility. In fact, dysphagia at the last visit was a predictor of shorter survival. However, by contrast with what has been suggested in progressive supranuclear palsy, early dysphagia was not a predictor of shorter survival.14Indeed, this lack of predictive power may be related to a differentially later onset of dysphagia in corticobasal degeneration.
Diagnostic accuracy remained poor among primary neurologists, less than a half of the patients were diagnosed before death. Even retrospective blinded evaluation of the clinical features in 10 of these 14 patients by movement disorders specialists disclosed poor sensitivity.4 This low sensitivity may be explained in part by the absence of a distinctive deficit in some cases. Previous reports1 emphasised the fixed dystonic jerky and apraxic arm as highly suggestive of corticobasal degeneration. Once these symptoms are found specificity is 100%.4 However, the present case series suggests that less severe presentations such as unilateral limb bradykinesia with mild ideomotor apraxia and without superimposed dystonia or myoclonus should provide a major index of suspicion, not only in early disease but also at advanced stages of corticobasal degeneration. The additional presence of focal cognitive involvement should raise the possibility of corticobasal degeneration.
Additional laboratory investigations that may support the diagnosis of corticobasal degeneration, but were not systematically evaluated in our study, include: distinct asymmetry of sulci in the parietal region on CT or MRI15 16; asymmetry of cortical and subcortical fluorodeoxyglucose or oxygen metabolism on PET17-20; perfusion asymmetry of parietal cortical regions on HMPAO SPECT21; reduced basal ganglia IBZM uptake on SPECT22; decreased horizontal saccade latency on oculography23; short latency reflex myoclonus; absence of back averaged cortical potentials preceding the action myoclonus; or abnormal magnetic stimulation of the motor cortex.24 25Clinical studies may include disorders that can simulate features of corticobasal degeneration such as Alzheimer’s disease, progressive supranuclear palsy, Pick’s disease, hemiatrophy-parkinsonism, and multiple system atrophy of striatonigral degeneration type.4 These diseases may share clinical features with corticobasal degeneration making them indistinguishable in life from corticobasal degeneration confirmed at postmortem examination. Thus at present, postmortem examination remains the ultimate diagnostic gold standard. On the other hand, studies confirmed by postmortem examination may include more atypical cases. The ideal study, unfortunately hard to implement at the present time, would include postmortem confirmed prospectively followed up community cases of corticobasal degeneration.
Acknowledgments
We thank Drs S Daniel, D Dickson, DS Horoupian, and PL Lantos for providing cases for the NINDS database. We also thank DG Schoenberg for editing the manuscript.