Article Text

Abstract
A family is described in which one member presented with symptoms and signs suggestive of corticobasal degeneration and a sibling presented with features of a frontal lobe dementia. Their mother developed a presenile dementia and movement disorder. At postmortem examination the member with clinical corticobasal degeneration had non-specific pathological features. Therefore, the clinical features of corticobasal degeneration can occur with non-specific pathological changes. Within a pedigree, different members can present with different clinical syndromes, which may reflect variation in the distribution and severity of the pathological process.
- frontotemporal dementia
- corticobasal degeneration
Statistics from Altmetric.com
Some degenerative diseases can present with the combination of a movement disorder and a dementia including Huntington’s disease, progressive supranuclear palsy, and diffuse Lewy body disease. By contrast, the neurodegenerative dementias such as Alzheimer’s disease, Pick’s disease, and frontotemporal dementia present with cognitive symptoms and motor problems are usually late, or minor, manifestations. We present clinical details on three patients within a pedigree. Two presented with a movement disorder and the third with a frontal lobe dementia. The proband, who presented with features of corticobasal degeneration, lacked specific pathological features of corticobasal degeneration or other neurodegenerative disease on postmortem examination.
Methods
CLINICAL
The family was referred to the Dementia Research Group through an unaffected younger sibling. The two affected patients in the second generation were examined.
PATHOLOGICAL
Blocks of tissue were taken from the frontal, temporal, parietal and occipital lobes, basal ganglia, thalamus with the subthalamic area, cerebellar vermis and hemisphere, midbrain, pons, and medulla oblongata. Altogether 20 blocks of tissue were examined.
Histological stains included haematoxylin and eosin, luxol fast blue, and cresyl violet and modified Bielschowsky silver impregnation.
Immunocytochemistry using the avidin-biotin complex method was carried out using antibodies to ubiquitin, tau protein, glial fibrillary acidic protein (all Dako), α-β-crystallin (Novacastra), β-amyloid, and SP 40 prion protein (both provided by Professor BH Anderton).
Results
CLINICAL
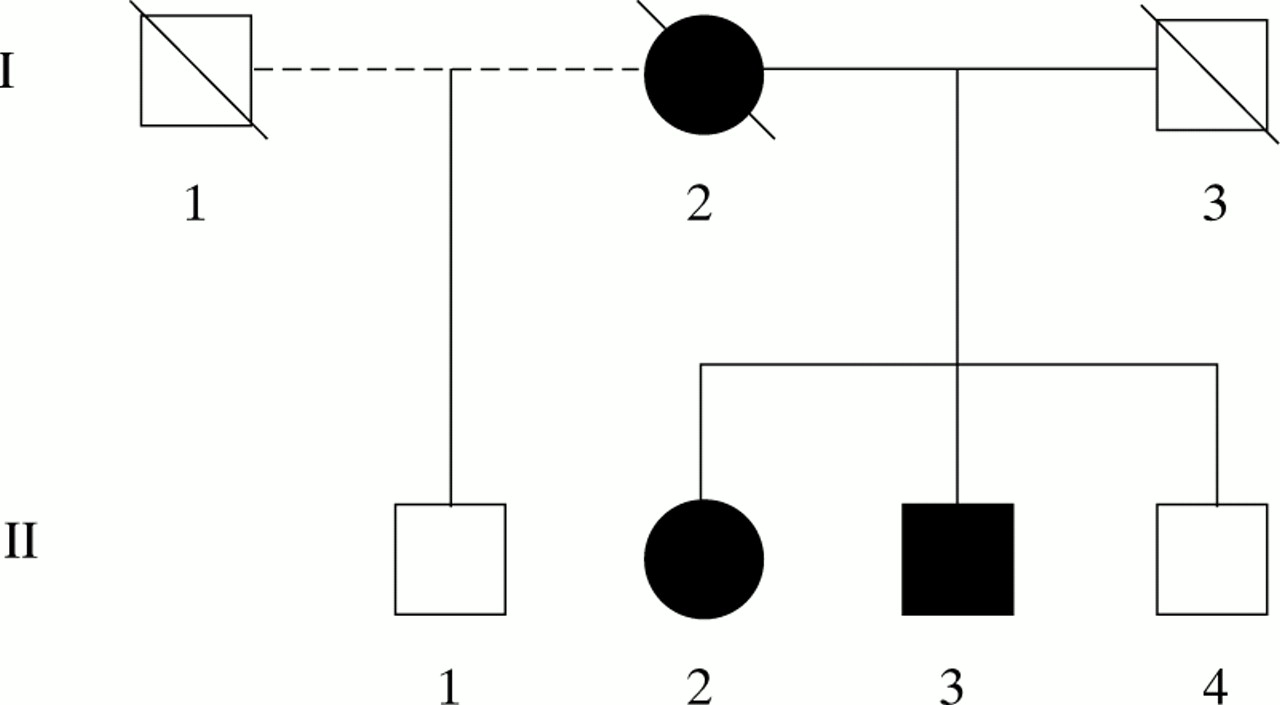
The pedigree is shown in figure 1.
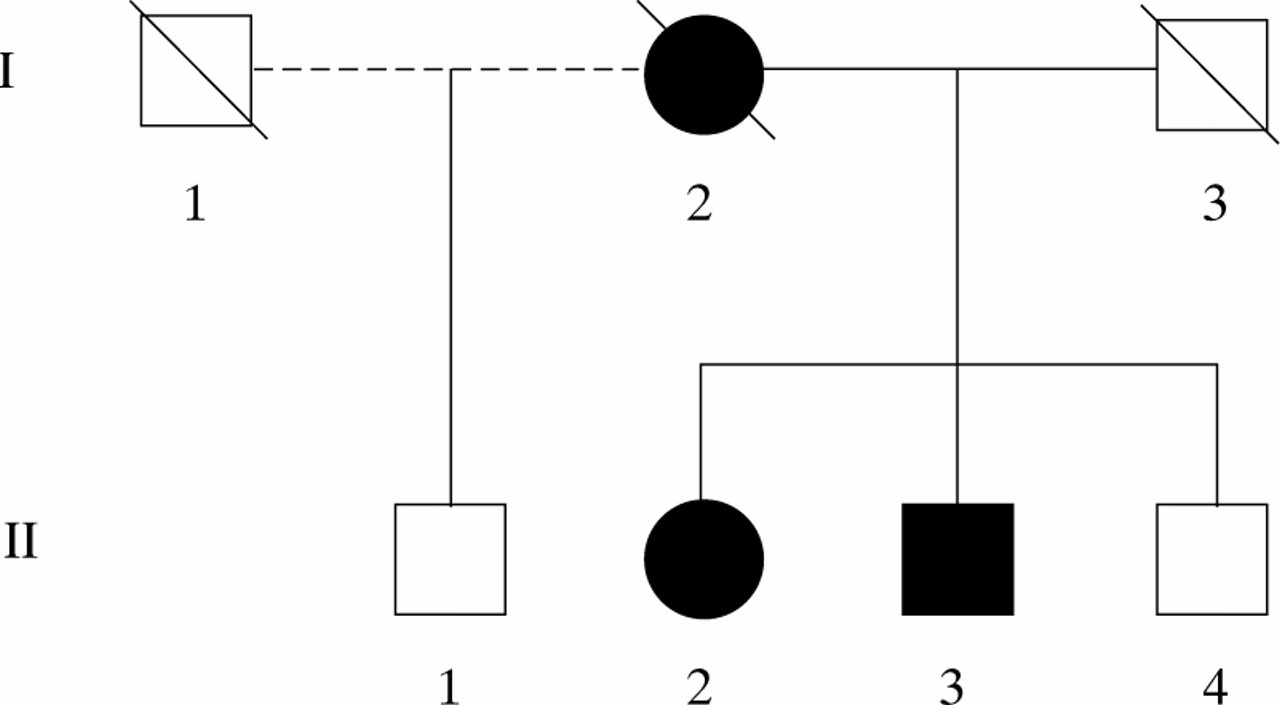
The pedigree.
I.1, male—No information available
I.2, female—A housewife who developed a left hemiplegia in her fifth decade. After this, she had a progressive memory disturbance—for example, she started hanging her coat in the pantry and forgetting to put a kettle on the lighted gas. She became aggressive and wandered, deteriorating steadily before becoming bed bound. She died at the age of 54 years.
I.3, male—No information available.
II.1, male—No cognitive problems at the age of 64 years.
II.2, female—A right handed dressmaker with a history of hypercholesterolaemia. At the age of 64 she complained of a postural tremor and clumsiness of her left hand. A few weeks later, she started to drag her left leg. Her motor symptoms gradually progressed and she developed urinary incontinence. She became distractible, often leaving taps running. She began to wander, often becoming lost. She was tearful and emotionally labile and kept asking “Have I got what my mother had?”.
She was admitted at the age of 64 for full assessment to the National Hospital for Neurology and Neurosurgery, Queen Square. On examination, she had marked impersistence of both eye closure and gaze. She had a mild left upper motor neuron facial weakness, facial hypomimia, and a reduced blink rate. She was dysarthric with an exaggerated jaw jerk and brisk pout reflex. Her gait was wide based and unsteady. Her left hand was held stiff, in a dystonic posture with a marked, unilateral, fast frequency, postural tremor. She had markedly increased tone in her left arm with some cogwheel rigidity. A grasp reflex could be elicited in her left hand. Tone was mildly increased in the other limbs. There was secondary akinesia of left hand and arm movements. She had a mild left sided weakness in a pyramidal distribution. Reflexes were symmetrically brisk but plantar responses were flexor. No alien limb phenomenon was seen. The only abnormal sensory finding was dysgraphaesthesia in her left hand. She had bilateral apraxia for copying hand postures and miming gestures and could not copy sequential or alternating hand movements. Her hand writing was small and illegible. Brain CT showed mild asymmetric cerebral atrophy more marked on the right. An EEG showed a preserved alpha rhythm. Routine tests including syphilis serology and CSF examination were unremarkable and a neurophysiological examination was normal.
She was disinhibited and tried to cheat on the psychometric tests. She scored 30/30 on the mini mental state examination (MMSE).1Psychometry showed a verbal IQ of 97 and a performance IQ of 79 (Wechsler adult intelligence scale - revised (WAIS-R)). The National adult reading test (NART) suggested that her premorbid IQ was high average. Her reading was good. She scored well on the McKenna object naming test. Spelling was normal. She had no dyscalculia. Her performance on visuospatial and perceptual tests was adequate. She could sort the Weigl shapes. Her verbal fluency was reduced to seven words beginning with “S” in a minute. Her proverb interpretations were concrete and personalised. She could describe a picture adequately with prompting. Card sorting was performed slowly but accurately. A clinical diagnosis of probable corticobasal degeneration was made.
The next year she was reassessed. She had developed marked emotional lability and had become aggressive towards her husband. She had delusions that strangers were in her house. She had great difficulty initiating walking and her gait was very unsteady. She also had difficulty chewing and swallowing food. On examination she had a pseudobulbar palsy with a tremor of her tongue. She had oral rooting to approaching visual stimuli. Her left hand was held stiff and useless. Plantar responses remained flexor. She now scored 23/30 on the MMSE, scoring well on orientation, registration, and recall and moderately well on calculation. No perceptual problems were detected. She recalled details of the last visit. Verbal fluency was reduced to a single word beginning with “S” in a minute. While reading aloud she developed a progressive hypophonia. She gradually deteriorated and died a year later.
NEUROPATHOLOGY
The fixed brain weighed 955 g; the brainstem and cerebellum weighed 144 g.
MACROSCOPY
There was severe atrophy of the frontal, parietal, and temporal lobes in this order of severity. The occipital lobes were relatively well preserved. The atrophy was more severe on the right: the right frontal and parietal lobes were most severely affected. The leptomeninges were slightly thickened. The cranial nerves were normal. The large cerebral arteries had a few atherosclerotic plaques with narrowing of their lumina up to 25%. On coronal slicing the lateral ventricles were greatly enlarged with rounding of their angle. The gyri were narrowed and the intervening sulci widened. There was additional space between the hippocampus and the wall of the inferior horn of the lateral ventricle on both sides and the cortical ribbon was narrowed. The right side was more severely affected and the frontoparietal regions were most severely involved. The substantia nigra and the locus coeruleus were paler than usual. No focal lesions were seen.
MICROSCOPY
There was superficial laminar status spongiosus in the frontal, temporal, parietal, and insular cerebral cortices with neuronal loss and patchy astrocytosis. In the hippocampus there was neuronal loss in the CA1 area with increased glial fibres, pigment containing macrophages, and Hirano bodies, but neurofibrillary tangles and the senile plaques here, as in the neocortex, were extremely rare. The neurofibrillary tangles, demonstrated both by ubiquitin and tau protein, were mainly in the superficial cortical layers. Here, there were a few ubiquitin positive neuropil threads. In the frontal and parietal cortex an occasional swollen neuron was seen.
There was mild neuronal loss and astrocytosis in the caudate nucleus. Vacuole formation with astrocytosis was seen in the globus pallidus, whereas the putamen was better preserved. The nucleus basalis of Meynert was well populated, but there was extraneuronal pigment and an occasional faintly fibrillar inclusion in the locus coeruleus and substantia nigra; these appeared to be tau positive. The substantia nigra also showed moderately severe neuronal loss and astrocytosis. There was axonal swelling of the Purkinje cells in the cerebellum; the dentate was normal. The white matter showed moderate astrocytosis. There was some fibrous thickening of blood vessel walls including some calcified material in the basal ganglia.
Immunocytochemistry disclosed minimal β-amyloid deposition. Immunostaining for prion protein was negative.
II.3, male—An electrician. At the age of 57 years he started having arguments at work and making errors on account of which he retired early. Later that year, while on holiday, he became lost and was found 5 days later exhausted and bedraggled, many miles away. He lacked insight into his illness and became increasingly distractible. He developed repetitive behaviour, for example taking up to 16 walks a day; as he walked he would say aloud the names of roads he passed. He began speaking to strangers and became less kempt.
He was examined at the age of 63 when he was found to have a positive pout reflex and a visual rooting reflex. His blink rate was reduced. Otherwise, general neurological examination was normal. By contrast his behaviour was clearly abnormal. Thus he joked incessantly—for example: “My wife’s broomstick failed the MOT”. He repeatedly and spontaneously gave the time to the minute, and constantly walked around an internal square counting the windows and touching each one as he went past. He often swore, just audibly, and repeatedly said “bitch” and “I’ll strangle her”. He scored 21/30 on the MMSE1 scoring well on calculation, visuospatial skills, and spatial orientation. He had poor concentration and was facetious. His interpretation of proverbs was concrete, cognitive estimates were adequate. On testing verbal fluency, he named five complex words such as “somnambulist” in a minute. Spontaneous speech was limited and perseverative and his picture description lacked fluency. By contrast he scored 10/10 on the Queen Square screening test for cognitive deficits naming test.2 He read a passage well and his spelling was normal. He had mild apraxia for miming gestures and could not learn a motor sequence or alternating hand movements. He perseverated on a card sorting test. Routine blood tests were normal. Brain CT showed cerebral atrophy, particularly of the anterior two thirds of the brain.
II.4, male—No cognitive problems at the age of 60 years.
Discussion
Patient II.2 presented with a gradual onset, progressive asymmetric motor syndrome starting in the arm and spreading to the leg. She developed an asymmetric akinetic rigid syndrome, ocular impersistence, cortical sensory loss, asymmetric postural tremor, dysequilibrium, and a focal dystonia. These are all typical features of corticobasal degeneration.3 4 She developed the rigid, dystonic, jerking, and completely useless left hand which Lang et al thought might be unique to corticobasal degeneration.4 Three of her clinical features could be regarded as atypical for classic corticobasal degeneration: the rapid progression of her symptoms to other limbs, the absence of an alien limb phenomenon, and her early cognitive problems. The rapid spread of her symptoms is a striking feature of this case but does not suggest an alternative diagnosis. The alien limb phenomenon is not required for a clinical diagnosis of corticobasal degeneration and is absent in 40% of described cases.4 Her early cognitive problems were dominated by distractibility with a perfect score on the MMSE suggesting a predominantly frontal lobe syndrome. As her memory, while she remained ambulant, was well preserved her cognitive problems would not exclude her from accepted criteria for corticobasal degeneration.4 Furthermore an increasingly wide range of clinical features has now been recognised in the disease.5Therefore, a clinical diagnosis of corticobasal degeneration was made, although she was not regarded as a classic case. Only brief clinical details are available for her mother (I.2) but it seems that she had a similar illness. Corticobasal degeneration has recently been reported to occur on a familial basis.6
By contrast, her brother (II.3) presented with a frontal lobe dementia with personality change, pathological joking, facetiousness, and stereotyped routines. He fulfilled the clinical criteria for frontotemporal dementia.7 His late, mild extrapyramidal features are consistent with this diagnosis.7
Clinically diagnosed corticobasal degeneration normally has a distinctive pathological picture.3 Macroscopically there is usually asymmetric atrophy of the frontal, parietal, and peri-Rolandic cortex, pallor of the pigmented nuclei of the brainstem, and a variable degree of ventricular enlargement. Microscopy shows swollen cortical neurons, tau positive neuronal and glial (chiefly astrocytic) inclusions, and corticobasal inclusions in the degenerated substantia nigra, in addition to neuronal loss and astrocytosis of the affected areas. A recent symposium reviewed corticobasal degeneration and its relation to other asymmetric cortical degeneration syndromes and recognised variation in the pathological features in patients clinically diagnosed as corticobasal degeneration8—for example, those lacking swollen achromatic neurons and with no significant basal ganglia pathology.9 The disease has also been reported in association with Alzheimer’s pathology with occasional swollen neurons10 and with Alzheimer’s pathology with Lewy bodies.11
Our patient lacked significant numbers of swollen cells and the more obvious nigral pathology associated with corticobasal degeneration although there were occasional tau positive fibrillary inclusions in both the locus coeruleus and substantia nigra. Moreover, the extensive tau pathology and glial pathology typical of corticobasal degeneration were lacking. Therefore, the disease can be associated with non-specific pathological changes as well as with classic corticobasal pathology and Alzheimer’s pathology. This variability in the pathological findings is well known in frontotemporal dementia and other neurodegenerative syndromes. Patients with a predominantly frontal lobe syndrome usually lack specific pathological changes at postmortem examination, but can have features of Pick’s disease,12 Alzheimer’s disease,13corticobasal degeneration,13 or motor neuron disease.14 Figure 2 summarises the current pathological correlates of these clinical syndromes.

{kind=link}
{kind=link}
Clinical neurodegenerative syndromes and their known pathological correlates.
The other novel feature in this family is the occurrence of clinical corticobasal degeneration and frontotemporal dementia in those within the same pedigree. The strikingly different clinical presentations may reflect variation in the distribution and severity of the pathological process. Such variation in the clinical picture despite a common pathological process is well known in other neurological diseases such as the GM2 gangliosidoses and familial prion disease. Identification of the common pathological process in this family would significantly advance our understanding of neurodegenerative disease. Molecular genetic techniques offer the most obvious route to this goal.
Acknowledgments
JB was supported by the MRC and Action Research. We are grateful to Miss Penelope Roques for her help in tracing the family. The help of Dr NJ Cairns, Mrs Susan Wise, and Mr A Chadwick, members of the MRC London Neurodegenerative diseases Brain Bank, Department of Neuropathology, Institute of Psychiatry, is gratefully acknowledged.